Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(13):7495-7513. doi:10.7150/thno.131907 This issue Cite
Research Paper
CDK9 degrader induces BRCAness and sensitizes castration-resistant prostate cancer to PARP inhibitor
Jiaxuan Li1,2#, Jingya Sun3#, Weisong Tan4#, Guoqi Li5, Wenjie Xiao3,6, Jiajun Wu7, Wei Yang8, Changqing Chen1, Yang Li1, Jiakuan Liu1, Yuanyu Liu4, Dong Liu1, Xiao-hua Chen3,8, Rujian Zhu1 ![]() , Ruimin Huang3,5,6
, Ruimin Huang3,5,6 ![]() , Jun Yan1
, Jun Yan1 ![]()
1. Department of Urology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center; Laboratory Animal Center, Fudan University, Shanghai 200032, China.
2. Shanghai Institute of Infectious Disease and Biosecurity, School of Public Health, Fudan University, Shanghai 200032, China.
3. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China.
4. Laboratory Animal Center, Fudan University, Shanghai 200032, China.
5. School of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China.
6. University of Chinese Academy of Sciences, Beijing 100049, China.
7. State Key Laboratory of Genetic Engineering, School of Life Sciences, Fudan University, Shanghai 200438, China.
8. School of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China.
# These authors contributed equally to this article.
Received 2026-1-21; Accepted 2026-5-24; Published 2026-6-17
Abstract

Rationale: Castration-resistant prostate cancer (CRPC) poses significant therapeutic challenges due to its aggressive nature and limited effective treatments. Although PARP inhibitor olaparib has been approved for metastatic CRPC patients bearing BRCA1/2 mutations, its application is confined to this specific patient subpopulation. The induction of “BRCAness” feature in CRPC patients without BRCA1/2 mutations becomes a significant challenge.
Methods: A transcriptomic analysis to identify potential “BRCAness” regulator was performed on 921 prostate cancer (PCa) patients from 6 public datasets and validated in our own cohort. D45, a selective small-molecule protein degrader for cyclin-dependent kinase 9 (CDK9), alone or combined with olaparib, was applied in CRPC cell lines (C4-2 and 22Rv1) lacking BRCA1/2 mutations for cell viability, colony formation and apoptosis-related assays. “BRCAness” phenotype was characterized by Western blotting and γH2AX foci accumulation assays. RNA-seq and CUT&Tag assays were used to reveal how D45 regulated homologous recombination repair (HRR)-related genes.
Results: We found the transcriptional regulator CDK9 was overexpressed in CRPC and correlated with advanced Gleason scores, metastasis, and poor prognosis. D45 treatment decreased cell survival, and led to downregulation of HRR-related genes (BRCA1/2 and RAD51) with the reduced recruitment of phosphorylated-RNA polymerase II (pSer2) to the ends of these genes with increased DNA damage, indicating a “BRCAness” phenotype induction. The synthetic vulnerability synergized with D45 plus olaparib was thus tested in vitro, showing the enhanced apoptosis in CRPC cells. Moreover, sequential D45 and olaparib administration significantly suppressed 22Rv1 xenograft growth in vivo (P < 0.001), reduced RAD51 expression and increased DNA damage. Toxicity was tolerable and consistent with prior reports. Their synergistic effect was confirmed in ex vivo explants from human PCa specimens.
Conclusions: Our findings demonstrated CDK9 as a master regulator of BRCAness and proposed targeting CDK9 as a potential strategy to sensitize CRPC patients without BRCA1/2 mutations to PARP inhibition.
Keywords: castration-resistant prostate cancer, BRCAness, CDK9 protein degrader, PARP inhibitor, homologous recombination repair
Introduction
Prostate cancer (PCa) remains the most prevalent malignancy and one of the leading causes of cancer-related death in men worldwide, with castration-resistant prostate cancer (CRPC) representing the lethal form driven by persistent androgen receptor (AR) signaling and adaptive survival mechanisms [1-3]. About 84% of CRPC patients advance to metastatic stage, while conventional treatments for metastatic CRPC (mCRPC) patients often prove inadequate, resulting in a 5-year survival rate of only ~30% [4]. In light of these challenges, development of novel and more effective therapeutic strategies for CRPC is urgent.
Next-generation sequencing reveals that ~25% of mCRPC patients harbor homologous recombination repair (HRR) gene defects, including BRCA2 (8.7%), ATM (5.9%), and BRCA1 (1-6.3%) mutations, conferring “BRCAness” vulnerability to poly (ADP-ribose) polymerase (PARP) inhibitor (PARPi) via synthetic lethality [5-9], which occurs between two specific genes when the perturbation of either gene alone is viable but the perturbation of both genes simultaneously leads to cell death [10, 11]. PARPi could prevent DNA repair, resulting in the accumulation of DNA damage. Olaparib, as a well-known PARPi, has already been approved for HRR or BRCA1/2 gene-mutated mCRPC by FDA, EMA and NMPA in the period 2020-2021 [12, 13]; and rucaparib, another PARPi, was also approved by FDA for BRCA-mutated mCRPC in 2020 [14]. Even though PARPis have improved progression-free survival in biomarker-selected cohorts, their clinical utility remains constrained: only 12-20% of CRPC patients exhibit BRCA1/2 alterations, and acquired resistance via HRR restoration frequently limits therapeutic durability [8]. These results reveal the following limitations of PARPi application on CRPC in clinic: 1) it is only applicable in patient harboring specific genetic defects; 2) single-agent of PARPi has limited efficacy; 3) acquired resistance has occurred inevitably. To overcome these barriers, strategies inducing homologous recombination deficiency have emerged to sensitize BRCA1/2-proficient tumors [15, 16]. Targeting molecular drivers that regulate both CRPC progression and HRR competency could expand PARPi efficacy to broader populations. This approach aims to leverage pharmacological induction of BRCAness, bypassing reliance on pre-existing genetic defects while mitigating resistance mechanisms, thereby redefining precision oncology paradigms for mCRPC.
Cyclin-dependent kinases (CDKs), a family of serine/threonine kinases, orchestrate tumor progression through cell cycle regulation, transcriptional control, and metabolic reprogramming. CDK9, a pivotal transcription-related kinase, drives RNA polymerase II (RNA Pol II)-mediated oncogene expression by phosphorylating the C-terminal domain, a rate-limiting step in transcriptional elongation [17, 18]. Elevated expression of CDK9 was found in different types of cancer, such as leukemia, breast cancer and PCa. CDK9 promotes cancer progression mainly through controlling the transcription of MYC and MCL1, two important genes for cancer cell apoptosis and survival [19, 20]. Thus, CDK9 has become a promising target for anticancer therapy, and strategies for blocking CDK9 transcriptional activity have been developed. For example, small-molecule inhibitors of CDK9 were designed to occupy its ATP-binding pocket [21, 22]. Pharmacological CDK9 inhibition exhibited broad anticancer potential against renal carcinoma and PCa cells [23, 24]. However, some CDK9 inhibitors, such as flavopiridol, suffered from dose-limiting toxicities due to off-target CDK inhibition [25]. Benefited from proteolysis targeting chimeras (PROTACs) technique, the development of CDKs protein degraders indicates a promising therapeutic strategy for treating transcription-addicted cancers [26]. For example, a PROTAC BSJ-5-63 (targeting CDK12, CDK7 and CDK9) could diminish BRCA1/BRCA2 expression and attenuate AR signaling, sensitizing CRPC to PARP inhibitors and enhancing the therapeutic efficacy [27]. Whether these effects are dependent on CDK9 and how CDK9 is involved in “BRCAness” induction, need to be thoroughly investigated in mCRPC without HRR gene defects.
We previously developed a potent and selective CDK9 degrader (D45), which could induce degradation of CDK9 protein, and lead to apoptosis in vitro along with xenograft growth inhibition in triple-negative breast cancer [28]. In this study, to address the significant challenge in CRPC patients without BRCA1/2 mutations, i.e., how to induce “BRCAness” state to sensitize patients to PARPi therapy, D45 treatment was tested, showing the “BRCAness” with the downregulation of HRR-genes, including BRCA1/2, PALB2, and RAD51. A sequential regimen of D45 and olaparib was applied in CRPC xenografts in vivo and human PCa explants ex vivo, and demonstrated the pre-existing DNA damage caused by CDK9 degrader was exacerbated by PARP inhibition, resulting in the accumulation of unrepaired DNA double-strand breaks and ultimate cell death.
Materials and Methods
Public data analysis
TCGA-PRAD dataset was obtained from The Cancer Genome Atlas Program (TCGA) database, with clinical information retrieved from cBioPortal (https://www.cbioportal.org). DKFZ-PRAD dataset was sourced from the Prostate Cancer dataset published in Cancer Cell 2018 [29] on cBioPortal; human PCa-related datasets (GSE35988, GSE70768, GSE8511, and GSE74367) and CDK9 inhibitor or degrader treatment-related datasets (GSE205363, GSE244716, GSE169090, and GSE282602) were downloaded from the Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo). For analysis on mRNA expression level in TCGA-PRAD dataset, log2(FPKM+1) value of each probe was applied, and average value was used for the genes with multiple probes. The human PCa proteomic datasets OEP004998 and PXD049446 were obtained from the National Omics Data Encyclopedia (https://www.biosino.org/node) and ProteomeCentral (https://proteomecentral.proteomexchange.org/), respectively.
PCa cell lines and human prostate specimens
Human PCa cell lines 22Rv1 and C4-2 cells were purchased from American Type Culture Collection. Cells were maintained in RPMI1640 medium, supplemented with 10% fetal bovine serum (FBS; ThermoFisher Scientific, USA) and penicillin (100 U/mL) plus streptomycin (100 μg/mL) at 37 °C in 5% CO2. Human benign prostatic hyperplasia (BPH), PCa and CRPC specimens were obtained from Shanghai Pudong Hospital. All procedures were performed in compliance with relevant laws and institutional guidelines and have been approved by Ethics Committees of Shanghai Pudong Hospital, Fudan University Pudong Medical Center (2025-KY-019-E01 and 2026-IIT-012-E01). The informed consent was obtained for experimentation with human subjects.
Small interfering RNA (siRNA) transfection
siRNAs targeting human CDKs and non-specific control siRNA were purchased from Sangon Biotech (China). Following the manufacturer’s protocol, C4-2 cells were plated in 6-well plates at 2 × 106 cells per well or in 96-well plates at 3 × 103 cells per well, and transfected with 40 nM of siRNA duplex using Lipofectamine RNAiMAX (13778150, ThermoFisher Scientific) in Opti-MEM (31985070, ThermoFisher Scientific). 48 h after transfection, cells were used for further experiments. The siRNA sequences were listed in Table S1.
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences (2020-09-HRM-40 and 2024-06-HRM-79). For spontaneous CRPC model, both PbCre;PtenF/F and control PtenF/F mice were maintained on C57BL/6 genetic background and underwent orchiectomy at 4-5 months of age. After 12 months, these mice were euthanized, and the prostate tissues were harvested for histological analysis.
For the fragment implantation of CRPC xenograft model, 1 × 107 22Rv1 cells were injected subcutaneously on the flanks of 6 to 8-week-old male BALB/c nude mice (GemPharmatech, China). The original subcutaneous tumors were harvested upon reaching a volume of 500 mm3, and chopped into small blocks (~ 2 mm3) for further subcutaneous implantation via trocar into 6-week-old male BALB/c nude mice. Once the xenograft volume reached 70 mm3, 1.5 mg/kg of D45 and/or 50 mg/kg of olaparib (HY-10162, MedChemExpress, China) were intraperitoneally (ip) administrated according to the indicated schedule. For the combination treatment group, D45 was injected first, and olaparib was injected 6 h later; single drug or vehicle (PBS) were used as the control groups. The body weight of the mice was recorded and the xenograft volume was measured with a caliper daily. Tumor volumes were calculated using the formula: Volume = (length × width2) × 0.5. Two weeks later, the animals were euthanized and the xenografts were harvested for further analyses.
For drug toxicity assessment, 8-week-old male C57BL/6 mice received 1.5 mg/kg of D45 and/or 50 mg/kg of olaparib via ip administration according to the indicated schedule for 28 consecutive days. For the combination treatment group, D45 was administered first, followed by olaparib after a 6 h-interval; and control groups received either the single agent or vehicle. Body weight and clinical signs were recorded daily. At the end of the 28-day treatment period, mice were sacrificed, and peripheral blood was collected for complete blood count (CBC) using a hematology analyzer (XN-1000V, Sysmex, Japan) and for serum biochemical analysis using a biochemical analyzer (Cobas C501, Roche, Germany) with the corresponding kits for alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CREA), and urea. Major organs (heart, liver, lung, kidney, small intestine, and spleen) were harvested for histopathological assessment via hematoxylin and eosin (H&E) staining.
Immunohistochemistry (IHC) staining
Tissues were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at a thickness of 5 μm. The slides were deparaffinized in xylene, rehydrated through graded alcohol and underwent antigen retrieval in citrate buffer (pH 6.0) for 30 min. Endogenous peroxidase activity was blocked with a peroxidase reagent for 10 min, and non-specific binding was blocked with normal goat serum for 10 min (KIT-9707, MXB Biotechnologies, China). Sections were incubated with primary antibodies at 4 °C overnight, followed by the corresponding secondary antibodies at room temperature for 30 min. 3,3'-diaminobenzidine (DAB; MXB Biotechnologies) was used for visualization with hematoxylin counterstaining. The antibody information was listed in Table S2.
Western blotting assay
Total protein lysates were obtained using RIPA lysis buffer containing protease and phosphatase inhibitors (M5293 and M7528, Abmole, China). 20 µg protein lysates were separated by SDS-PAGE and transferred to 0.45 μm nitrocellulose membranes (Merck Millipore, USA), followed by blocking with TBST containing either 5% skimmed milk powder or 5% bovine serum albumin (A600332, Sangon Biotech) at room temperature for 1 h. After the incubation with primary antibodies at 4 °C overnight, the secondary antibody was incubated at room temperature for 1 h. The signal was detected using the enhanced ECL chemiluminescent substrate (Shanghai Tanon Life Science, China), and the images were captured using the ChemiDoc MP Imaging System (Bio-Rad, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted by Trizol (15596018, ThermoFisher Scientific) and cDNA was synthesized using Hifair AdvanceFast 1st Strand cDNA Synthesis Kit (11149ES60, YEASEN, China). qPCR was further performed using ChamQ Universal SYBR qPCR Master Mix (Q311-02, Vazyme, China). mRNA expression levels were normalized to ACTB, using the 2-ΔΔCt method to calculate the relative expression levels. The primer sequences were listed in Table S1.
Cell viability assay
22Rv1 and C4-2 cells were seeded in 96-well plates at a density of 3,000 cells/well. Cells were treated with D45 and/or olaparib for 72 h. MTT solution (5 mg/mL) was added and incubated at 37 °C for 4 h. Formazan crystals were dissolved with 100 μL DMSO per well, followed by an incubation at 37 °C for 15 min. Absorbance was measured at 490 nm using a microplate reader, with 680 nm as the reference. The combination index (CI) of D45 and olaparib was calculated using Compusyn software (ComboSyn, UK). The combined drug effects were quantified according to the CI theorem from Chou-Talalay’s method, which defines: additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1).
Colony formation assay
Cells were seeded into 12-well plates at a density of 1,500 cells/well. After 2-3 days, cells were treated with various concentrations of D45 and/or olaparib for 7 days. Colonies were subsequently fixed with 4% formaldehyde, washed with PBS and stained with 0.5% crystal violet solution (Sigma-Aldrich, USA).
Apoptosis analysis
Cells were seeded in 6-well plates at 1.5 × 105/well, and D45 and/or olaparib at the indicated concentrations were added on the next day for 72 h. Both floating and adherent cells in each well were collected, washed with PBS and stained with FITC-Annexin V in a buffer containing propidium iodide (PI) using FITC Annexin V Apoptosis Detection Kit I (556547, BD Biosciences, USA), followed by flow cytometry (NovoCyte, Agilent Technologies, USA). The Annexin V-positive cells were defined as apoptosis.
RNA-seq assay
22Rv1 cells were treated with D45 (40 nM) and vehicle for 12 h, respectively. Total RNA was isolated with Trizol according to the manufacturer's instructions. After measurement by 5300 Bioanalyser (Agilent Technologies) and quantification by NanoDrop 2000 spectrophotometer (ThermoFisher Scientific), 1 μg RNA (OD260/280 = 1.8~2.2, OD260/230 ≥ 2.0, RQN ≥ 6.5, 28S:18S ≥ 1.0) was used to generate sequencing library for RNA-seq (Majorbio Biotech, China).
Differentially-expressed gene (DEG) analysis was performed using the DESeq2. Genes responsive to CDK9 degraders were identified based on a threshold of P < 0.05 and |Fold Change| ≥ 1.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were conducted using DAVID platform (https://david.ncifcrf.gov). Gene Set Enrichment Analysis (GSEA) was performed based on expression matrices (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). HRR genes were identified using Human DNA Repair Genes database (https://www.mdanderson.org). Data were visualized by bubble plots generated with the R package, and heatmaps, Venn diagrams, and GO/KEGG pathway visualizations were created using Bioinformatics online tools (http://www.bioinformatics.com.cn). For Gene Set Variation Analysis (GSVA), the WP_HR signaling pathway from MSigDB was analyzed using the GSVA R package, and sample-specific scores were generated for subsequent analysis. Drug sensitivity prediction was conducted using the oncoPredict R package. The training dataset was obtained from Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org).
Immunofluorescence staining
Cells were seeded in 6-well plates on coverslips at a density of 1 × 105 cells/well and treated with D45, olaparib, or a combination for 12 h. For the knockdown experiment, after 24 h of siRNA treatment, olaparib was added for 12 h. Cells were fixed with ice-cold methanol, followed by blocking in 5% goat serum in the presence of 0.1% Triton X-100. The primary antibody was incubated at 4 ℃ overnight, and Alexa 488 or 555-conjugated secondary antibodies were incubated at room temperature for 1 h, respectively. Finally, the nuclei were stained with DAPI (P0131, Beyotime, China) and photographed under an Olympus FV3000 confocal laser scanning microscope.
CUT&Tag (Cleavage Under Targets & Tagmentation) assay
C4-2 cells were treated with 40 nM D45 or vehicle for 12 h and harvested for CUT&Tag assay using NovoNGS CUT&Tag 4.0 High-Sensitivity Kit (N259-YH01, Novoprotein, China). In brief, 1 × 105 cells were immobilized on concanavalin A beads and subsequently incubated with IgG or phospho-RNA Pol II CTD (C-terminal domain) (Ser2; 703108, ThermoFisher Scientific) as primary antibody for 2 h, and goat anti-rabbit (N269, Novoprotein) as the secondary antibody at room temperature for 1 h. These samples were then incubated with ChiTag transposon and fragmented with MgCl2. Genomic DNA was extracted and amplificated using the indexing primers. The enrichments on BRCA1, BRCA2 and RAD51 loci were measured by qPCR with the primers listed in Table S1.
DR-GFP reporter assay
HRR efficiency was assessed using the DR-GFP reporter system [30, 31]. Briefly, 293T cells were seeded into 6-well plates at a density of 2 × 105 cells per well and cultured overnight. Cells were then co-transfected with the DR-GFP reporter plasmid and an I-SceI endonuclease expression plasmid using Lipofectamine 3000 transfection reagent (L3000015, ThermoFisher Scientific) according to the manufacturer's instructions. At 24 h post-transfection, cells were exposed to D45 and/or olaparib at the indicated concentrations for an additional 24 h. Subsequently, the proportion of GFP-expressing cells was quantified by flow cytometry (Agilent Technologies) and normalized to represent relative HRR efficiency.
Ex vivo explants from human PCa specimens
The culture of human PCa explant ex vivo was performed as described previously [32, 33]. Briefly, PCa tissues were cut into small fragments, put on the absorbable gelatin sponge (HSD-B, HUSHIDA Shanghai, China) and transferred into 6-well plates with 2 mL culture medium (DMEM/F-12 containing 10% FBS and penicillin plus streptomycin) per well at 37 °C in 5% CO2. The tissue fragments were treated with 20 nM D45 for 24 h, followed by 20 μM olaparib treatment for 48 h; treatments with vehicle or D45 alone (20 nM for 72 h) or olaparib alone (20 μM for 48 h) were used as controls. After treatment, tissue fragments were collected for histological assessment. Three PCa tissues were examined, including patient #1 (Gleason score 4+3/ISUP grade group 3), #2 (Gleason Score 4+3/ISUP grade group 3), and #3 (Gleason score 4+5/ISUP grade group 5).
Statistical analyses
Statistical analyses were conducted using Prism 9.5.0 (GraphPad Software, USA). Continuous variables between two groups were compared by unpaired Student’s t-test. Multi-group comparisons were analyzed via one-way ANOVA (for one independent variable) or two-way ANOVA (for two independent variables), followed by Tukey’s post hoc test. Kaplan-Meier survival curves were generated, and intergroup differences were assessed by log-rank test. Pairwise correlations were quantified by Pearson correlation coefficient. P value < 0.05 was defined statistical significance.
Results
CDK9 overexpression in CRPC patients was associated with poor prognosis
To investigate whether CDK family member was correlated with CRPC, the integrated bioinformatic analyses were performed across three independent cohorts (TCGA-PRAD, GSE35988, and GSE70768) using the mRNA levels of 21 CDK members and the information on CRPC status or biochemical recurrence (BCR). CDK1, 4, 9, 10, and 16 were consistently upregulated in CRPC patients or PCa patients with BCR in at least two datasets (Figure 1A). In C4-2 CRPC cells, siRNA knockdown of these five CDKs (Figure S1A-E) revealed that depleting CDK9 decreased cell viability more potently than depleting the other four (Figure 1B). Moreover, CDK9 overexpression was observed in human PCa patients from TCGA-PRAD (P < 0.001; Figure 1C), which was significantly correlated with advanced Gleason score (P < 0.05; Figure 1D-E), T stage (P < 0.001; Figure S1F-G), metastatic progression (P < 0.01; Figure S1H-I) and recurred/progressed status (P < 0.001; Figure S1J) in TCGA-PRAD, DKFZ-PRAD, and GSE8511 datasets. Notably, PCa patients with BCR (TCGA-PRAD) or CRPC patients (GSE35988, GSE70768 and GSE74367) exhibited higher CDK9 expression levels than those without BCR (P < 0.01; Figure 1F) or non-CRPC patients (P < 0.05; Figure 1G-H and Figure S1K), portending poor patient survival (P < 0.05 in TCGA-PRAD and DKFZ-PRAD) (Figure 1I-J).
CDK9 was overexpressed in CRPC. A, The heatmap of CDK family members in three public PCa datasets. Color-coded blocks, the ratio of their expression levels between CRPC and non-CRPC (GSE35988 and GSE70768) or between BCR+ and BCR- PCa samples (TCGA-PRAD) with P < 0.05; gray blocks, P ≥ 0.05 (ns); black blocks, mRNA expression levels unavailable (no probe). P values were calculated with the Limma-moderated t-test. B, Cell viability of C4-2 cells transfected with the indicated siRNAs after 48 h by MTT assay. NC, siRNA control. Two siRNAs targeting different regions of CDK family genes were tested. C, CDK9 mRNA levels in normal prostate and PCa tissues in TCGA-PRAD dataset. D-E, CDK9 mRNA levels in PCa patients with different Gleason scores (< 7 and ≥ 7) in TCGA-PRAD (D) and DKFZ-PRAD (E) datasets. F, CDK9 mRNA levels in PCa patients with and without BCR in TCGA-PRAD dataset. G, CDK9 mRNA levels in normal, localized PCa (LPCa) and metastatic CRPC (mCRPC) samples in GSE35988 dataset. H, CDK9 mRNA levels in normal, PCa and CRPC samples in GSE70768 dataset. I-J, The Kaplan–Meier plot of disease-free survival (I) and recurrence-free survival (J) in PCa patients, stratified by CDK9 mRNA levels in TCGA-PRAD (I) and DKFZ-PRAD (J) datasets, respectively. K-L, IHC staining of CDK9 protein in human BPH, localized PCa (LPCa) and CRPC tissues from our cohort (K), with the quantification of CDK9 protein among three groups (L). M-N, IHC staining of CDK9 protein in aged matched PtenF/F prostate tissues (control, CTL), naïve PbCre;PtenF/F PCa (CRPC-) and castrated PbCre;PtenF/F PCa (CRPC+) (M), with the quantification of CDK9 protein among three groups (N). Scale bar, 50 μm and 10 μm (inset). Data were represented as mean ± SD. P values were determined by Student's t-test in C-F, and one-way ANOVA test in B, G, H, L, N. *P < 0.05, **P < 0.01, ***P < 0.001, ns, P ≥ 0.05.
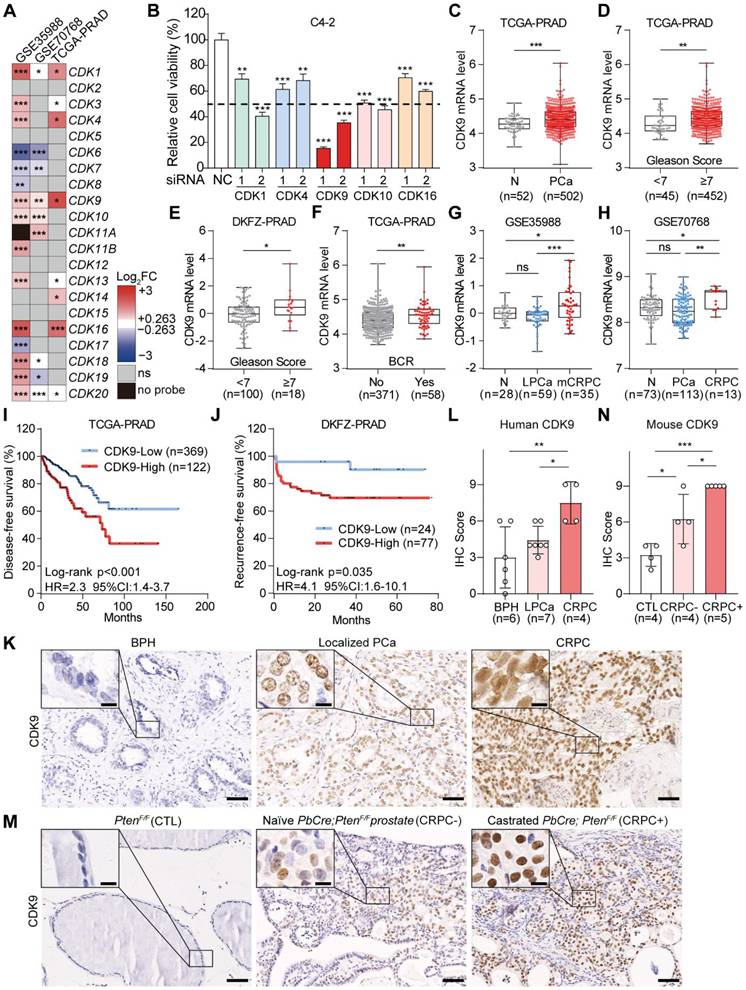
In line with the above results from public datasets, Western blotting data from our own cohort showed that both two isoforms of CDK9 proteins, due to different transcriptional start site [34], were significantly upregulated in PCa samples (n = 12), comparing with BPH samples (n = 12) (P < 0.001; Figure S1L-M). IHC staining further validated the marked overexpression of CDK9 protein in the nuclei of CRPC cells (n = 4), compared to naïve PCa (P < 0.05; n = 7); and BPH specimens (n = 6) showed the weakest CDK9 protein levels (Figure 1K-L). Murine PbCre;PtenF/F CRPC models recapitulated this CDK9 overexpression pattern, i.e., weak expression in PtenF/F prostate epithelial cells, moderate in PbCre;PtenF/F PCa cells, and strong in PbCre;PtenF/F CRPC cells (P < 0.05; Figure 1M-N). Collectively, CDK9 was significantly associated with CRPC, indicating its role as a potential target for PCa aggressive progression.
D45 decreased CDK9 protein levels in CRPC cells
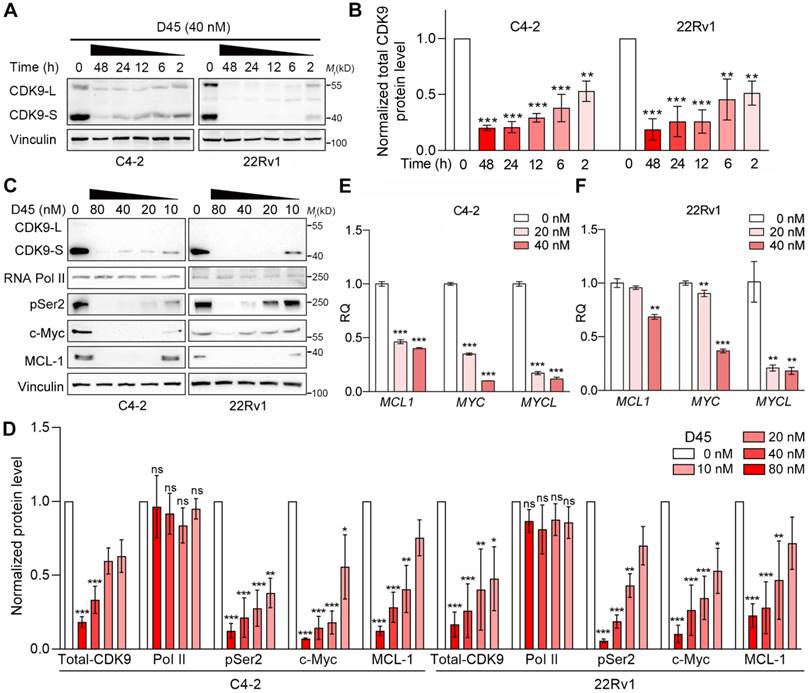
Compound D45, a PROTAC previously shown to induce CDK9 degradation and suppress xenograft growth in triple-negative breast cancer [28], was evaluated for CDK9-targeting efficacy in two CRPC cell lines, C4-2 and 22Rv1 [35]. Concomitant degradation of both CDK9 isoforms, the full-length CDK9-L and the shorter CDK9-S, was induced by D45 in a time- and dose-dependent manner (Figure 2A-D). CDK9 ablation by D45 could suppress the phosphorylation of RNA Pol II at serine 2 (pSer2) - a hallmark of transcriptional elongation - concomitant with the reduced expression of two canonical CDK9 downstream targets (MYC and MCL1) at both transcriptional and protein levels. Another MYC-related gene, MYCL, was also downregulated at mRNA level by D45 treatment (Figure 2C-F). These findings showed that D45 was a potent CDK9 degrader in CRPC contexts, effectively disrupting CDK9-mediated transcriptional addiction via suppression of its downstream oncogenic networks.
CDK9 degradation by D45 treatment in CRPC cells. A-B, Representative images (A) and quantification (B) for Western blots of CDK9 protein levels in C4-2 and 22Rv1 cells with 40 nM D45 at the indicated timepoints. CDK9-L, full-length isoform; CDK9-S, the short isoform. Total CDK9 protein levels (CDK9-L plus CDK9-S) were normalized by corresponding vinculin protein levels (n=3, mean ± SEM). C-D, Representative images (C) and quantification (D) for Western blots on protein levels of CDK9 and its downstream targets in C4-2 and 22Rv1 cells treated with various concentrations of D45 for 8 h. Vinculin was used as the normalization control (n=3, mean ± SEM). E-F, The mRNA levels of CDK9’s target genes in CRPC cells treated with D45 (E, C4-2 for 6 h; F, 22Rv1 for 12 h) by qRT-PCR assay (n=3, mean ± SD). P values determined by one-way ANOVA test in B, D, E, F. *P < 0.05, **P < 0.01, ***P < 0.001, ns, P ≥ 0.05.
D45 decreased the viability of CRPC cells in vitro
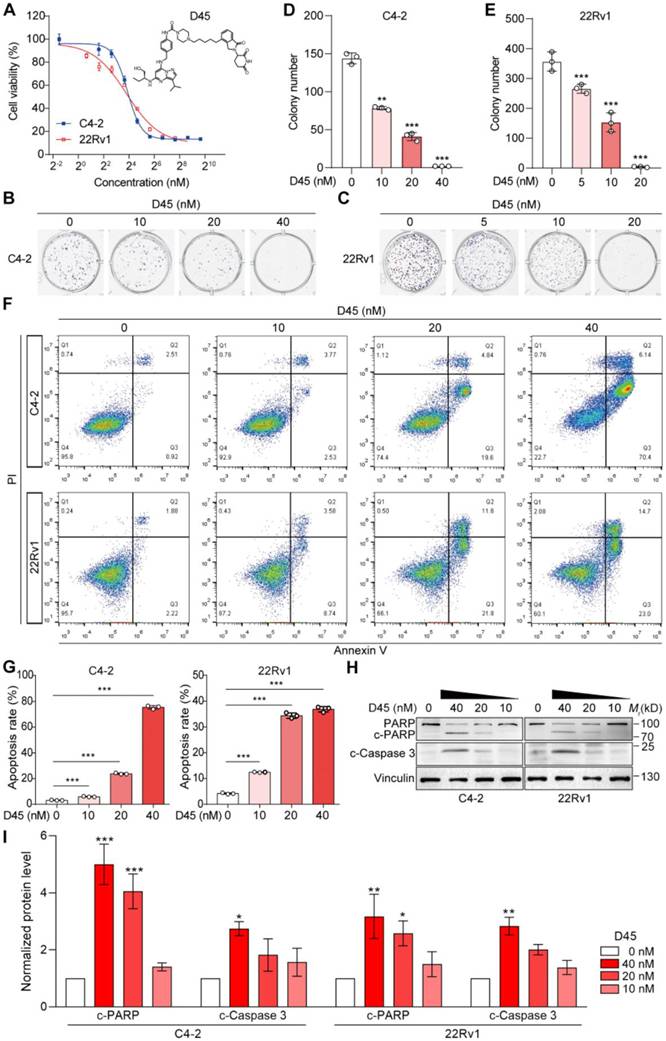
To assess the cytotoxic effects of D45 on CRPC cells, we conducted cell viability and colony formation assays using C4-2 and 22Rv1 cells. The results showed that both CRPC cells were sensitive to D45, with IC50 values of 14.70 nM for C4-2 and 13.99 nM for 22Rv1 (Figure 3A). D45 inhibited the colony formation capability of C4-2 and 22Rv1 cells in a dose-dependent manner (P < 0.01; Figure 3B-E). Flow cytometry analyses confirmed the dose-responsive apoptosis in CRPC cells to D45 (P < 0.001; Figure 3F-G), corroborated by cleavages of Caspase 3 and PARP, two apoptotic markers (Figure 3H-I). These results indicated that D45 effectively decreased the cell viability and enhanced apoptosis in CRPC cells.
D45 decreased the viability of CRPC cells in vitro. A, Cell viability of C4-2 and 22Rv1 cells treated with D45 at the indicated concentrations for 72 h by MTT assay. B-C, The colony formation capability of C4-2 (B) and 22Rv1 cells (C) treated with D45 at the indicated concentration for 7 days. D-E, Quantification of the colony numbers in B and C, respectively. F, Apoptosis in C4-2 and 22Rv1 cells treated with D45 at the indicated concentrations for 72 h with the Annexin V/PI staining by flow cytometry. G, Quantification for the apoptosis percentage in C4-2 and 22Rv1 cells in F. H-I, Representative images (H) and quantification (I) for Western blots of cleaved Caspase 3 (c-Caspase 3) and cleaved PARP (c-PARP) proteins in C4-2 and 22Rv1 cells treated with D45 at the indicated concentrations for 72 h (n=3). Vinculin was used as the normalization control. Data were represented as mean ± SD in D, E, G or mean ± SEM in I. P values were determined by one-way ANOVA test in D, E, G, I. *P < 0.05, **P < 0.01, ***P < 0.001.
D45 inhibited the HRR gene expression in CRPC cells
To delineate the mechanism underlying D45-induced cytotoxicity, transcriptomic profiling of D45-treated 22Rv1 cells (40 nM vs vehicle) was performed to reveal the CDK9-dependent transcriptional regulation network (GSE288031). Transcriptomic analysis detected a total of 41,016 gene transcripts. Among these, 4,283 genes were downregulated (10.4%) and 4,052 were upregulated (9.9%), while the majority (32,681 genes) showed no significant change. These data indicated that CDK9 degradation did not induce global transcriptional suppression. GO and KEGG analyses on DEGs demonstrated the coordinated suppression of DNA repair, double-strand break repair, and homologous recombination, which was regulated by HRR signaling (such as BRCA1/2, FANCF, PALB2, RAD51D, and XRCC2; Figure 4A-C), a pattern conserved across different datasets involving CDK9 inhibitors/degraders against various PCa cells (degrader LL-K9-3 to 22Rv1 cells in GSE205363 [24]; inhibitor AT7519 to LNCaP cells in GSE169090 [36] and inhibitor CDKI-73 to LNCaP cells in GSE244716 [37]; Figure S2A-B), as well as breast cancer cells (degrader compound 28 to MDA-MB-231 cells in GSE282602 [28]; Figure S2C-D). In addition, GSVA on the human PCa dataset containing CRPC status information (GSE35988, n = 94) linked the elevated CDK9 expression to HRR activation, including the significant upregulation of BLM, BRCA1, FANCE, RAD52, and XRCC2, along with the increase of WP_Score for HRR signaling pathway (r > 0.2, P < 0.05, 95% Confidence Interval (CI): 0.03-0.41; Figure S3).
Targeting CDK9 by D45 inhibited HRR gene expression in CRPC cells. A, Heatmap depicting the DEGs with |Fold Change| ≥ 1.5 in 22Rv1 cells following 12 h-treatment with 40 nM D45 or vehicle (Veh) by RNA-seq. B-C, GO (B) and KEGG (C) enrichment analyses of DEGs from RNA-seq data in (A). D-E, The representative images of γH2AX foci in C4-2 cells treated with D45 for 12 h by immunofluorescence staining (D) and the percentage of cells that had more than 10 γH2AX foci per cell (E). DAPI was used for nuclei staining. Scale bar, 10 μm. F-G, mRNA levels of HRR genes in C4-2 (F) and 22Rv1 cells (G) treated with D45 at the indicated concentrations by qRT-PCR assay. H-I, Representative images (H) and quantification (I) for Western blots of HRR signaling proteins in C4-2 and 22Rv1 cells treated with D45 at the indicated concentrations (n=3). β-Actin was used as the normalization control. J, CUT&Tag-qPCR analysis of the indicated HRR genes by phospho-RNA Pol II CTD (Ser2) antibody (POLR2A) in C4-2 cells treated with 40 nM D45 or vehicle (Veh). IgG was used as a negative control. Data were represented as mean ± SD (in E, F, G, J) or mean ± SEM (in I). P values were determined by Hypergeometric test in B, C, by one-way ANOVA test in E, F, G, I, and Student's t-test in J. *P < 0.05, **P < 0.01, ***P < 0.001.
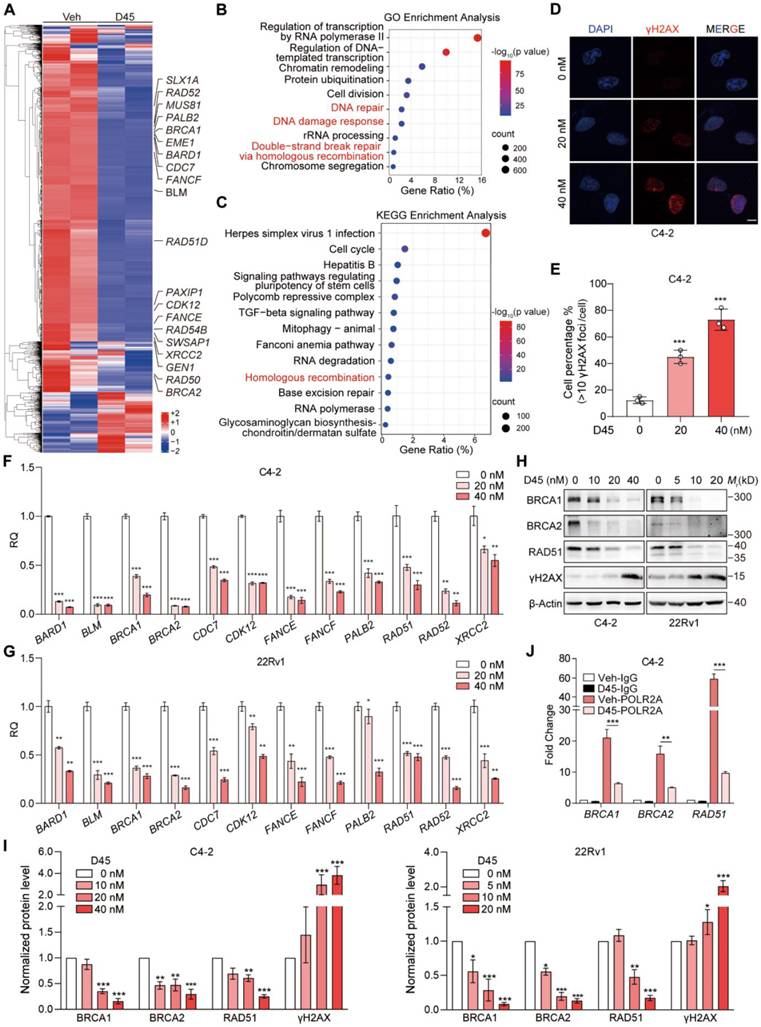
Functional validation showed that D45 could induce γH2AX foci (DNA damage markers; Figure 4D-E), and decrease the mRNA (Figure 4F-G) and protein levels (Figure 4H-I) of HRR-related genes in C4-2 and 22Rv1 cells in a dose-dependent manner. Inhibiting CDK9 with siRNAs mimicked these results (Figure S4A-C). The CTD of human RNA Polymerase II Subunit A (POLR2A) comprises 52 tandem consensus heptapeptide repeats (Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7). pSer2 within these repeats is primarily regulated by CDK9 to facilitate transcriptional elongation [18, 38]. By analyzing public proteomics datasets, we confirmed that pSer2 levels were significantly elevated at multiple positions in human PCas compared to corresponding normal prostate tissues (Figure S5A). Furthermore, mean pSer2 levels were positively correlated with both CDK9 protein abundance (r = 0.63, P < 0.001, 95% CI: 0.43-0.77 in OEP004998 dataset [39]; r = 0.21, P = 0.0035, 95% CI: 0.069-0.33 in PXD049446 dataset [40]) and downstream HRR-related proteins, such as RAD50 (r = 0.50, P = 0.0002, 95% CI: 0.26-0.59 in OEP004998 dataset; r = 0.57, P < 0.0001, 95% CI: 0.48-0.66 in PXD049446 dataset) (Figure S5B). CUT&Tag assay was thus performed [41], revealing that D45 inhibited the enrichment of pSer2-RNA Pol II at the ends of key HRR genes, including BRCA1/2 and RAD51 (Figure 4J). These findings suggested that the CDK9/pSer2 axis drove HRR activation by enhancing transcriptional elongation of essential repair genes, indicating CDK9 as the potential target to generate BRCAness.
D45 synergized with olaparib to decrease CRPC cell viability and enhance DNA damage
Considering that targeting CDK9 downregulated a battery of HRR genes, including BRCA1/2 and RAD51, and exhibited BRCAness, drug sensitivity analysis was performed in GSE35988 and found the predicted IC50 values of olaparib were significantly higher in CRPC patients than those in non-CRPC PCa (P < 0.001; Figure S6A). Those values were also positively correlated with CDK9 mRNA levels (P < 0.01; Figure S6B). The combination of CDK9 depletion and PARP inhibition was thus suggested for CRPC treatment.
In C4-2 and 22Rv1 cells, olaparib alone decreased cell viability only to ~ 50% even at the highest concentration (40 μM for C4-2 and 80 μM for 22Rv1). In the presence of D45, olaparib (10 μM) plus D45 (10 nM) in C4-2 cells (Figure 5A) or plus D45 (5 nM) in 22Rv1 cells (Figure S7A) achieved > 50% inhibitory effects. In order to explore whether it is a synergistic effect, Chou-Talalay analysis was performed. The resulting CI values were less than 1 for most drug-combination groups (Figure 5B, Figure S7B), indicating CDK9 degrader-olaparib synergy in both cells. The dose combinations of D45: olaparib (20 nM: 20 μM) for C4-2 cells and D45: olaparib (10 nM: 20 μM) for 22Rv1 cells were therefore chosen for further studies. This drug combination significantly inhibited the colony formation capability (P < 0.001; Figure 5C-D, Figure S7C-D) and enhanced apoptosis (P < 0.01; Figure 5E-F, Figure S7E-F) in both cells. Western blotting assay confirmed the increased PARP cleavage and the decreased MCL-1 protein level (Figure 5G-H, Figure S7G-H). Additionally, this drug combination significantly reduced the BRCA1 foci (P < 0.001; Figure 6A-B) and elevated the γH2AX foci (P < 0.05; Figure 6C-D) by immunofluorescence staining in C4-2 cells. Western blotting assay verified the decrease of BRCA1/2 proteins and the increase of γH2AX protein in both C4-2 and 22Rv1 cells (Figure 6E-F). Moreover, the DR-GFP reporter assay indicated that D45, both as a monotherapy and in combination with olaparib, significantly impaired HRR efficiency (P < 0.001), whereas olaparib alone had no significant effect in 293T cells (Figure 6G-H). These effects were consistent with the observations using CDK9 siRNAs with olaparib (Figure S8A-D), further validating that decrease of CDK9 expression by either CDK9 degrader or CDK9 siRNA synergized with olaparib to exacerbate DNA damage by suppressing the HRR pathway.
D45 synergized with olaparib to decrease CRPC cell viability in C4-2 cells. A, Cell viability of C4-2 cells treated with D45 and olaparib, alone or combined, at the indicated concentrations for 72 h by MTT assay. B, Combination index for D45 and olaparib in C4-2 cells by Chou-Talalay analysis. C, Colony formation capability of C4-2 cells treated with D45 and olaparib, alone or combined, at the indicated concentrations. D, Quantification of colony numbers in C. E, Apoptosis in C4-2 cells treated with D45 and olaparib, alone or combined, at the indicated concentrations for 72 h with the Annexin V/PI staining by flow cytometry. F, Quantification for the apoptosis percentage in E. G-H, Representative images (G) and quantification (H) for Western blots of cleaved PARP (c-PARP) and MCL-1 protein in C4-2 cells treated with D45 and olaparib, alone or combined, at the indicated concentrations for 72 h. Vinculin was used as the normalization control (n=3). Data were represented as mean ± SD in D, F, or mean ± SEM in H. P values were determined by two-way ANOVA test in D, F, and by one-way ANOVA test in H. *P < 0.05, **P < 0.01, ***P < 0.001.
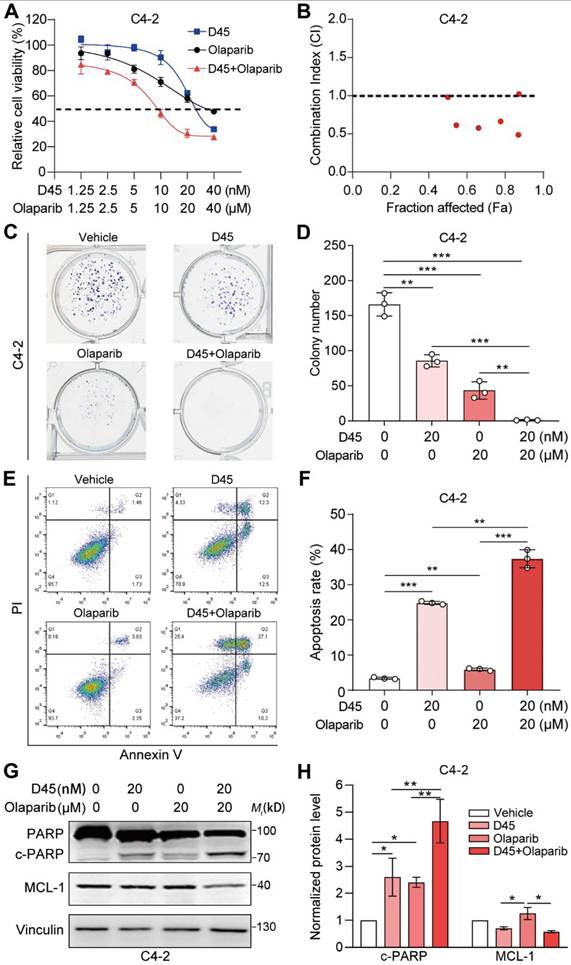
D45 synergized with olaparib to enhance DNA damage. A, C, Representative images of BRCA1 (A) and γH2AX foci (C) in C4-2 cells treated with D45 and olaparib, alone or combined, for 12 h by immunofluorescence staining. DAPI was used for nuclei staining. Scale bar, 10 μm. B, D, The percentage of cells that had more than 10 BRCA1 foci per cell in (A) and more than 10 γH2AX foci per cell in (C) (mean ± SD). E-F, Representative images (E) and quantification (F) for Western blots of BRCA1, BRCA2 and γH2AX proteins in C4-2 and 22Rv1 cells treated with D45 and olaparib, alone or combined, for 72 h (n=3, mean ± SEM). β-Actin was used as the normalization control. G-H, DR-GFP reporter assay evaluating the effects of D45 and olaparib on HRR efficiency in 293T cells (G). Relative HRR efficiency was normalized to the vehicle control (100%) (H). P values determined by two-way ANOVA test in B, D, H, and one-way ANOVA test in F. *P < 0.05, **P < 0.01, ***P < 0.001.
Sequential administration of D45 and olaparib significantly suppressed PCa xenograft growth in vivo
A 28-day repeated-dose toxicity study was conducted in C57BL/6 mice using the sequential dosing schedule (Figure S9A). Throughout the treatment period, no mortality or severe moribundity was observed, and body weight remained stable across all four groups (Figure S9B). Following the four-week administration, systemic safety was evaluated via CBC and serum biochemical analysis (Table S4). While the erythroid system exhibited moderate fluctuations, the most critical safety signal was a marked reduction in the leukocyte population. Specifically, white blood cell (WBC) and lymphocyte (LYMPH) counts decreased significantly in all three groups with drug treatment (P < 0.001); monocyte (MONO) and eosinophil (EO) counts were significantly reduced in the olaparib monotherapy (P < 0.05) and combination group (P < 0.01); a significant decline in neutrophil (NEUT) was observed exclusively in the combination group (P < 0.05). A myelosuppression was thus indicated, consistent with clinical observations of CDK9 inhibitor (dinaciclib) [42] and olaparib [43]. Whereas, serum biochemical analysis revealed no significant alterations in hepatic (ALT and AST) or renal (CREA and urea) function markers. Histopathological evaluation via H&E staining of major organs, including the heart, liver, lung, kidney, small intestine, and spleen, confirmed no significant morphological damage across all groups (Figure S9C).
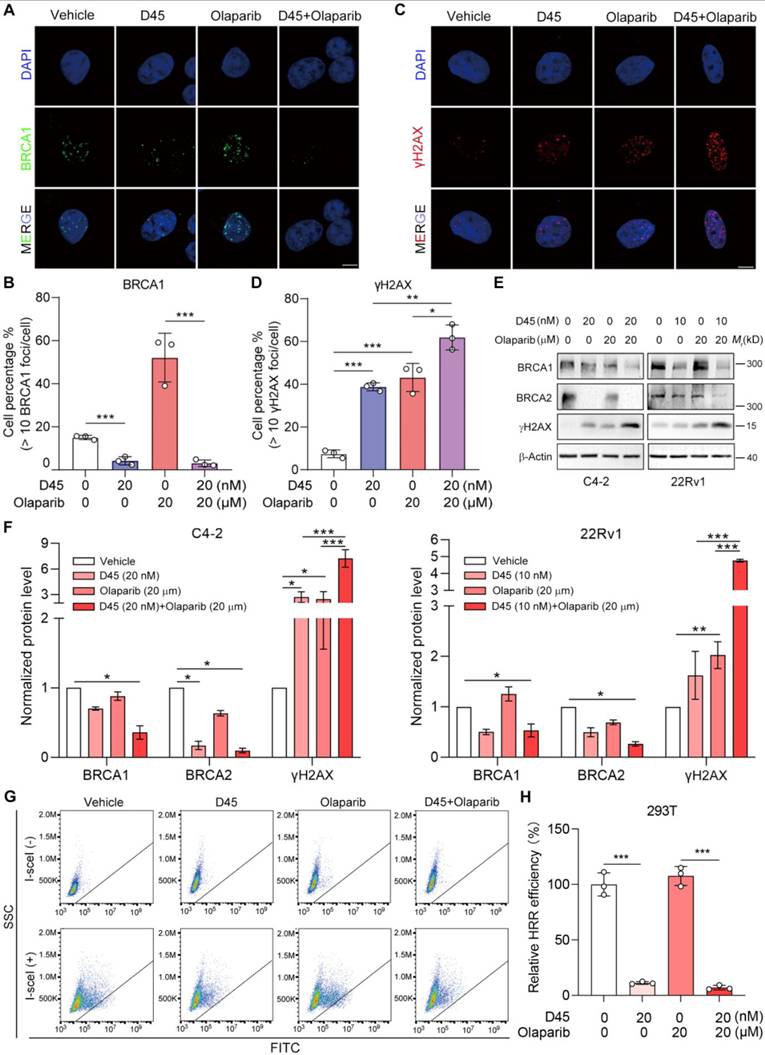
Regarding the tolerable toxicity profile, we established the subcutaneous 22Rv1 xenografts in nude mice to evaluate the efficacy of D45-olaparib combination in vivo. We implemented a sequential regimen to induce BRCAness status prior to PARPi treatment (Figure 7A): Low-dose D45 priming (1.5 mg/kg, ip, q.d. for first three days) was administered to induce the initial BRCAness, followed by intermittent combination therapy with D45 (1.5 mg/kg, ip, every other day) and olaparib (50 mg/kg, ip, 4×/week). After two weeks of combined administration, we found that D45 or olaparib alone had little effects compared to the vehicle group; however, drug combination significantly suppressed the xenograft growth comparing to the single drug group (P < 0.001; Figure 7B), without significant change in body weight (Figure 7C). At the endpoint, xenografts were harvested and weighed to confirm the tumor growth suppression (P < 0.01; Figure 7D-E). Western blotting assay demonstrated that combination treatment decreased CDK9 and RAD51 protein levels and increased PARP protein cleavage in 22Rv1 xenografts (Figure 7F-G). Consistently, IHC staining revealed that the combination treatment remarkably reduced RAD51 and Ki-67 (cell proliferation marker) protein levels, and enhanced γH2AX protein expression in 22Rv1 xenografts (Figure 7H-J).
Sequential administration of D45 and olaparib significantly suppressed 22Rv1 xenograft growth. A, Experimental scheme of sequential regimen for D45 and olaparib, alone or combined, in 22Rv1 xenografts. The xenograft-bearing mice were divided into four groups: Vehicle (n = 6), D45 (1.5 mg/kg, n = 6), olaparib (50 mg/kg, n = 6), and D45 plus olaparib (1.5 mg/kg and 50 mg/kg, n = 6). B-E, Tumor growth curve (B), body weight curve (C), tumor photo (D), and tumor weight (E) of 22Rv1 xenografts treated with D45 and olaparib, alone or combined. F-G, Representative images (F) and quantification (G) for Western blots of CDK9, RAD51, and cleaved PARP proteins in 22Rv1 xenografts treated with D45 and olaparib, alone or combined (n=3). Vinculin was used as the normalization control. H, The representative IHC images of Ki-67, γH2AX and RAD51 proteins in 22Rv1 xenografts treated with D45 and olaparib, alone or combined. Scale bar, 50 μm. I-J, Quantification of Ki-67 (I) and γH2AX proteins (J) in 22Rv1 xenografts treated with D45 and olaparib, alone or combined. Data were represented as mean ± SD in B-E, I-J, or mean ± SEM in G. P values were determined by two-way ANOVA test in B-E, I-J and by one-way ANOVA test in G. *P < 0.05, **P < 0.01, ***P < 0.001.
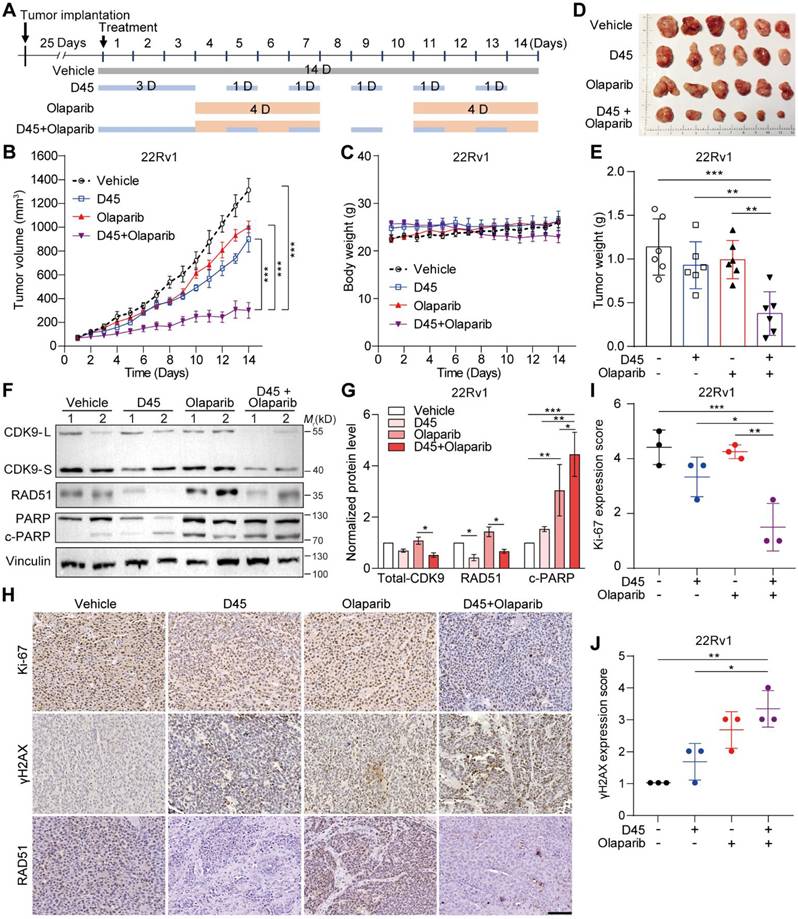
The combination regimen was further validated using ex vivo explants from human PCa tissues (Figure 8A-C). IHC analysis revealed that the combination therapy group showed a significant decrease in Ki-67 expression (P < 0.001; Figure 8D) and a significant increase in cleaved Caspase 3 (P < 0.001; Figure 8E), compared to the vehicle group and the single drug group, respectively.
Sequential treatment of D45 and olaparib significantly inhibited cell proliferation and induced apoptosis in human PCa explants ex vivo. A, Schematic diagram of the PCa explant culture process. B, Experimental schematic of PCa explants treated with D45 and olaparib, alone or combined. The PCa explants were divided into four groups (n = 3/group): Vehicle, D45 (20 nM), olaparib (20 μM), and D45 plus olaparib (20 nM and 20 μM). C, H&E staining for PCa explants treated with D45 and olaparib, alone or combined. Scale bar, 50 μm. D-E, IHC images and quantification for Ki-67 (D) and cleaved Caspase 3 proteins (E) in PCa explants treated with D45 and olaparib, alone or combined. Scale bar, 50 μm and 25 μm (inset). Data were represented as mean ± SD with P values determined by two-way ANOVA test in D, E. ***P < 0.001.
Taken together, the sequential treatment of D45-olaparib synergistically improved antitumor efficacy in vivo and ex vivo via suppressing HRR pathway, inducing DNA damage, and promoting apoptosis in PCa.
Discussion
In this study, we identified that CDK9 expression level was significantly elevated in CRPC patients and served as a poor prognosis in PCa patients, based on the analyses of 921 PCa patients from 6 public datasets, followed by the validation in our own cohort. Moreover, a CDK9 selective degrader (D45) was proved to efficiently reduce CDK9 protein level, inhibit the transcription of HRR genes, and induce “BRCAness” in CRPC cells. Notably, a sequential, low-dose regimen of D45 and olaparib achieved synthetic lethality and overcame PARPi resistance with minimal toxicity.
The clinical success of PARPi has established a paradigm for synthetic lethality in cancer treatment, particularly in tumors harboring BRCA1/2 mutations [44]. However, the emergence of acquired resistance has significantly limited their long-term efficacy. To overcome these challenges and expand the utility of PARPi beyond patients with HRR-deficient tumors, combination therapeutic strategies have become imperative.
For BRCA1/2-deficient PCa, tumors can evade PARPi-induced synthetic lethality through various mechanisms, including the restoration of HRR functionality via secondary mutations in key HRR genes [45] and loss of CHEK2 [46]. To counteract this, rational combinations have been explored. For instance, combination with AR inhibitors in CRPC not only targeted the primary oncogenic driver but also modulated the expression of DNA damage repair genes, thereby preventing HRR recovery and re-sensitizing tumors to PARPi [47]. However, BRCA1/2-deficient PCa patients are very limited because of the low mutation frequency. For BRCA1/2-proficient PCa (BRCAness-negative), a primary objective is to pharmacologically induce a BRCAness phenotype to confer their sensitivity to PARPi. This strategy involves exploiting vulnerabilities in PCa cells to create a synthetic lethal partnership with PARP inhibition. Promising approaches have been intensively explored, including targeting key nodes in the DNA damage response (DDR) network, such as ATR or depleting HRR-associated proteins (e.g. RAD51 and BRCA1/2). A multi-center Phase II study (NCT03787680) is testing the activity of olaparib and AZD6738 (an ATR inhibitor) in mCRPC (https://clinicaltrials.gov/study/NCT03787680). In addition, a peptide-based PROTAC drug targeting BRCA2, combined with PARP inhibitors to induce a synthetic lethal state, promoted cell death and induced tumor regression in PCa models [48]. In addition, targeting cell cycle regulators, such as CDK4 and CDK6 by CDK4/6i (palbociclib or abemaciclib) shows synergistic anti-CRPC cell viability with PARPi (olaparib) [49].
The combination of a CDK9 selective degrader (D45) and PARPi (olaparib) was investigated in CRPC models in vitro and in vivo. CDK9 inhibition or degradation alone has been demonstrated to suppress multiple oncogenic transcriptional and epigenetic pathways in PCa [37, 50], suggesting a potential therapeutic advantage over traditional treatments, particularly in patients with advanced disease [51]. However, first-generation CDK9 inhibitors operate through an occupancy-driven model by competing for the ATP-binding pocket to block kinase activity, which requires high drug concentrations to achieve efficacy and lacks sufficient selectivity, resulting in severe on- and off-target systemic toxicity that limits clinical applicability [20]. In contrast, CDK9 degraders operate via an event-driven (catalytic) mechanism, leveraging E3 ubiquitin ligase recruitment to induce the proteasomal degradation of the target protein. Beyond merely neutralizing enzymatic activity, we further addressed the critical non-kinase functions of CDK9. As a pivotal scaffolding protein, CDK9 maintains the structural integrity of the P-TEFb complex and other transcriptional regulatory assemblies [52]. In addition, CDK9 forms a specialized complex with Cyclin K (rather than Cyclin T1) which localizes to chromatin during replication stress. This complex is essential for stabilizing stalled replication forks and limiting the accumulation of single-stranded DNA, thereby facilitating recovery from replication arrest [53]. Therefore, considering that CDK9 degraders could comprehensively abrogate the protein’s enzymatic activity, scaffolding roles, and genome-protective mechanisms, we explored whether CDK9 degradation could serve as a combination partner with PARPi in CRPC models.
Our novel combination strategy included: 1) a CDK9 PROTAC (D45) was chosen for its high potency and high selectivity: It was shown that the IC50 value of D45 was 13.6 nM in MDA-MB-231 cells and at a concentration of 10 nM, D45 selectively degraded CDK9 without affecting CDK1, 2, 4, 5, 6 and 7 proteins [28]. We also confirmed its high potency as IC50 values below 15 nM in both CRPC cell lines. 2) the synergistic effect between D45 and olaparib was demonstrated: the CI values were less than 1 at most drug-combination groups in CRPC cells by Chou-Talalay analysis. D45-olaparib synergy allowed for the use of substantially lower, more therapeutically tolerable doses of each agent. For example, our dosing regimen of drug combination was D45 (1.5 mg/kg every other day) and olaparib (50 mg/kg four consecutive days each week) for two weeks. The growth of 22Rv1 xenografts was significantly suppressed, compared to vehicle or single agent (P < 0.001), without apparent body weight loss. When D45 alone was administered in MDA-MB-231 xenografts, the dosage (2.5 mg/kg every day for one week) resulted in a significant body weight loss at the 7th day (P < 0.01) [28]. Therefore, this dose-sparing effect is of critical importance, as it promises to mitigate toxicity profiles while maintaining robust anticancer efficacy and enhancing the potential therapeutic window for CDK9-based therapies. 3) the CDK9-PARP combination pair was demonstrated: D45-olaparib synergy was rooted in a logically sequential mode of action in vivo, i.e. D45 first acted to pharmacologically induce a BRCAness state in BRCA1/2-proficient CRPC cells, effectively creating an acquired vulnerability; olaparib then exploited this induced vulnerability by imposing synthetic lethality. This was a fundamental advance beyond the single-agent mechanism of CDK9 degrader, which primarily relies on transcriptional inhibition and the subsequent induction of apoptosis. This drug combination activated a second, powerful cytotoxic pathway: the pre-existing DNA damage caused by CDK9 degrader was exacerbated by PARP inhibition, leading to the accumulation of unrepaired DNA double-strand breaks, genomic instability and ultimate cell death. This dual-pathway activation accounted for the enhanced suppression of CRPC cell growth and apoptosis induction observed in our models. However, the clinical translation of CDK9-PARP targeting regimen must anticipate potential resistance landscapes. Beyond canonical PARPi resistance mechanisms, such as HRR restoration and SLFN11 loss [54, 55], and CDK9-specific adaptations including the L156F mutation [56] or ABCB1-mediated efflux [57], synergistic resistance may emerge through compensatory bypass signaling or epigenetic remodeling. For instance, cells might upregulate alternative transcriptional kinases (e.g., CDK12) or shift their survival reliance to the ATR-CHK1 axis to stabilize compromised replication forks; and epigenetic plasticity induced by CDK9 suppression could enable adaptive transcriptional rewiring. Deciphering these adaptive trajectories will be pivotal for designing intermittent dosing regimens to delay resistance onset or developing next-generation polytherapies to circumvent acquired resistance.
Unlike traditional transcriptional inhibitors, which are frequently constrained by short half-lives and the requirement for a high maximum concentration (Cmax) to maintain target occupancy, D45 exhibited a favorable pharmacokinetic profile with a prolonged half-life (17.34 h) and substantial systemic exposure (area under the plasma concentration-time curve (AUC) of 9,649 h·ng/mL following a 5 mg/kg i.v. dose) in mice [28]. Inherent interspecies differences necessitate rigorous Phase I dose-escalation studies to establish the optimal clinical exposure-response relationship in humans. Moreover, a four-week repeated-dose toxicity study evaluating our sequential dosing schedule revealed no mortality or moribundity, with body weights remaining stable across all experimental cohorts. While CBC, serum biochemical, and histopathological analyses confirmed the absence of significant structural damage to major organs, we observed moderate hematologic toxicity in the CDK9 degrader, olaparib, and combination groups, which was consistent with previous reports for these drug classes [42, 43]. Future optimization might prioritize refining intermittent dosing schedules to fully leverage the sustained pharmacodynamic effects of protein degradation while maximizing the therapeutic window.
Conclusions
CDK9 depletion represents a promising strategy to induce BRCAness and to expand the therapeutic use of PARPi in CRPC patients without HRR-gene mutations. Moving beyond the conventional use of CDK9 depletion as a standalone transcriptional blocker, we re-define it as a potent “sensitizing agent” that reprograms DNA repair capability of CRPC cells. Our findings advocate for a strategic shift in the use of CDK9-targeting compounds, highlighting their greatest potential not as monotherapies, but as rational partners in combination regimens designed to induce synthetic lethality and overcome the inherent limitations of PARPi in HRR-proficient cancers.
Abbreviations
PCa: Prostate cancer; CRPC: Castration-resistant prostate cancer; AR: Androgen receptor; mCRPC: Metastatic CRPC; HRR: Homologous recombination repair; PARP: Poly (ADP-ribose) polymerase; PARPi: PARP inhibitor; CDK: Cyclin-dependent kinase; RNA Pol II: RNA polymerase II; PROTAC: Proteolysis targeting chimera; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; BPH: Benign prostatic hyperplasia; siRNA: Small interfering RNA; IACUC: Institutional Animal Care and Use Committee; ip: Intraperitoneally; CBC: Complete blood count; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; CREA: Creatinine; H&E: Hematoxylin and eosin; DAB: 3,3'-diaminobenzidine; IHC: Immunohistochemistry; RT-qPCR: Reverse transcription-quantitative polymerase chain reaction; CI: Combination index; PI: Propidium iodide; DEG: Differentially-expressed gene; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; GSEA: Gene Set Enrichment Analysis; GSVA: Gene Set Variation Analysis; GDSC: Genomics of Drug Sensitivity in Cancer; CUT&Tag: Cleavage Under Targets & Tagmentation; CTD: C-terminal domain; BCR: Biochemical recurrence; pSer2: Phosphorylation of RNA Pol II at serine 2; 95% CI: 95% Confidence Interval; POLR2A: RNA Polymerase II Subunit A; WBC: White blood cell; LYMPH: Lymphocyte; MONO: Monocyte; EO: Eosinophil; NEUT: Neutrophil; DDR: DNA damage response; Cmax: maximum concentration; AUC: Area under the plasma concentration-time curve.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We greatly thank Dr. Lei Shi (Tianjin Medical University) for providing DR-GFP reporter system, Dr. Xin Li (Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences), Ms. Henglei Lu, Ms. Qirui Qiu, Ms. Siyuan Ren and Mr. Chuanxin Wang from Shanghai Institute of Materia Medica, Chinese Academy of Sciences for their technical supports. This study was supported by the National Natural Science Foundation of China (82473075 and 82103415), the Scientific Instrument Developing Project of the Chinese Academy of Sciences (PTYQ2026YZ0029), the Shanghai Leading Talent Program of Eastern Talent Plan (BJKJ2025018), the Project of Shanghai Municipal Health Commission (202140267), the Project of Key Medical Specialty and Treatment Center of Pudong Hospital of Fudan University (Zdzk2020-01), Talents Training Program of Shanghai Pudong Hospital (LJ202203), Outstanding Leaders Training Program of Pudong Health Committee of Shanghai (PWRl2023-04), the Healthcare Talents Youth Program of Shanghai Pudong New Area (2024PDWSYCQN-03), and the Talent Introduction Project of Shanghai Pudong Hospital (YJYJRC202302).
No artificial intelligence (AI) tools were used for image generation, data collection, data analysis, or figure preparation in this study. All experimental data, figures, and analyses were generated and validated by the authors.
Contributions
RH and JY designed the study. JL, JS, WT, GL, WX, JW, WY, and YYL performed experiments. CC, YL, JKL, DL, XC, RZ, RH, and JY provided experimental resources. JL, JS, WT, GL, and WX analyzed data. JL, JS, WT, GL, WX, RH, and JY wrote the manuscript. RZ, RH, and JY supervised research.
Data availability
The RNA-seq data were available in GEO database with the number GSE288031. Other data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xu J, Wang R, Guan X. Enhancing insights into Global Cancer Statistics 2022: implications for cancer control. Sci China Life Sci. 2025;68:294-6
2. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701-11
3. Siegel RL, Kratzer TB, Wagle NS, Sung H, Jemal A. Cancer statistics, 2026. CA Cancer J Clin. 2026;76:e70043
4. Olmos D, Lorente D, Alameda D, Cattrini C, Romero-Laorden N, Lozano R. et al. Treatment patterns and outcomes in metastatic castration-resistant prostate cancer patients with and without somatic or germline alterations in homologous recombination repair genes. Ann Oncol. 2024;35:458-72
5. Hussain M, Corcoran C, Sibilla C, Fizazi K, Saad F, Shore N. et al. Tumor Genomic Testing for >4,000 Men with Metastatic Castration-resistant Prostate Cancer in the Phase III Trial PROfound (Olaparib). Clin Cancer Res. 2022;28:1518-30
6. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S. et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2020;382:2091-102
7. Shui IM, Burcu M, Shao CX, Chen C, Liao CY, Jiang S. et al. Real-world prevalence of homologous recombination repair mutations in advanced prostate cancer: an analysis of two clinico-genomic databases. Prostate Cancer Prostatic Dis. 2024;27:728-35
8. Wei Y, Wu J, Gu W, Qin X, Dai B, Lin G. et al. Germline DNA Repair Gene Mutation Landscape in Chinese Prostate Cancer Patients. Eur Urol. 2019;76:280-3
9. Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pract. 2011;65:1180-92
10. O'Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18:613-23
11. Lahiri S, Hamilton G, Moore G, Goehring L, Huang TT, Jensen RB. et al. BRCA2 prevents PARPi-mediated PARP1 retention to protect RAD51 filaments. Nature. 2025;640:1103-11
12. Taylor AK, Kosoff D, Emamekhoo H, Lang JM, Kyriakopoulos CE. PARP inhibitors in metastatic prostate cancer. Front Oncol. 2023;13:1159557
13. Hommerding M, Hommerding O, Bernhardt M, Kreft T, Sanders C, Tischler V. et al. Real-world data on the prevalence of BRCA1/2 and HRR gene mutations in patients with primary and metastatic castration resistant prostate cancer. World J Urol. 2024;42:491
14. Anscher MS, Chang E, Gao X, Gong Y, Weinstock C, Bloomquist E. et al. FDA Approval Summary: Rucaparib for the Treatment of Patients with Deleterious BRCA-Mutated Metastatic Castrate-Resistant Prostate Cancer. Oncologist. 2021;26:139-46
15. Orhan E, Velazquez C, Tabet I, Fenou L, Rodier G, Orsetti B. et al. CDK inhibition results in pharmacologic BRCAness increasing sensitivity to olaparib in BRCA1-WT and olaparib resistant in Triple Negative Breast Cancer. Cancer Lett. 2024;589:216820
16. Li J, Zhi X, Chen S, Shen X, Chen C, Yuan L. et al. CDK9 inhibitor CDKI-73 is synergetic lethal with PARP inhibitor olaparib in BRCA1 wide-type ovarian cancer. Am J Cancer Res. 2020;10:1140-55
17. Bacon CW, D'Orso I. CDK9: a signaling hub for transcriptional control. Transcription. 2019;10:57-75
18. Egloff S. CDK9 keeps RNA polymerase II on track. Cell Mol Life Sci. 2021;78:5543-67
19. Mo C, Wei N, Li T, Ahmed Bhat M, Mohammadi M, Kuang C. CDK9 inhibitors for the treatment of solid tumors. Biochem Pharmacol. 2024;229:116470
20. Zhang Y, Shan L, Tang W, Ge Y, Li C, Zhang J. Recent Discovery and Development of Inhibitors that Target CDK9 and Their Therapeutic Indications. J Med Chem. 2024;67:5185-215
21. Wang WJ, Gao L, Wang S, Huang W, Meng XY, Hu H. et al. Discovery of Orally Bioavailable and Potent CDK9 Inhibitors for Targeting Transcription Regulation in Triple-Negative Breast Cancer. J Med Chem. 2024;67:10035-56
22. Shen YL, Wang YM, Zhang YX, Ma SJ, Yang LH, Zhao CG. et al. Targeting cyclin-dependent kinase 9 in cancer therapy. Acta Pharmacol Sin. 2022;43:1633-45
23. Kuang Z, Guo K, Cao Y, Jiang M, Wang C, Wu Q. et al. The novel CDK9 inhibitor, XPW1, alone and in combination with BRD4 inhibitor JQ1, for the treatment of clear cell renal cell carcinoma. Br J Cancer. 2023;129:1915-29
24. Li J, Liu T, Song Y, Wang M, Liu L, Zhu H. et al. Discovery of Small-Molecule Degraders of the CDK9-Cyclin T1 Complex for Targeting Transcriptional Addiction in Prostate Cancer. J Med Chem. 2022;65:11034-57
25. Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15:519-27
26. Nie HJ, Li BF, Sun J, Yuan Y, Zhang ZG, Hu H. et al. Discovery of a Potent, selective and orally bioavailable CDK9 degrader for targeting transcription regulation in Triple-Negative breast cancer. Bioorg Chem. 2024;153:107876
27. Gui F, Jiang B, Jiang J, He Z, Tsujino T, Takai T. et al. Acute BRCAness induction and AR pathway blockage through CDK12/7/9 degradation enhances PARP inhibitor sensitivity in prostate cancer. Sci Adv. 2025;11:eadu0847
28. Wei D, Wang HL, Zeng QH, Wang WJ, Hao BB, Feng XL. et al. Discovery of Potent and Selective CDK9 Degraders for Targeting Transcription Regulation in Triple-Negative Breast Cancer. J Med Chem. 2021;64:14822-47
29. Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y. et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell. 2018;34:996-1011 e8
30. Zhao J, Tian S, Guo Q, Bao K, Yu G, Wang X. et al. A PARylation-phosphorylation cascade promotes TOPBP1 loading and RPA-RAD51 exchange in homologous recombination. Mol Cell. 2022;82:2571-87 e9
31. Peng X, Huang X, Zhang S, Zhang N, Huang S, Wang Y. et al. Sequential Inhibition of PARP and BET as a Rational Therapeutic Strategy for Glioblastoma. Adv Sci (Weinh). 2024;11:e2307747
32. Dong Y, Liu J, Xue Z, Sun J, Huang Z, Jing Y. et al. Pao Pereira extract suppresses benign prostatic hyperplasia by inhibiting inflammation-associated NFkappaB signaling. BMC Complement Med Ther. 2020;20:150
33. Schiewer MJ, Goodwin JF, Han S, Brenner JC, Augello MA, Dean JL. et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134-49
34. Liu H, Herrmann CH. Differential localization and expression of the Cdk9 42k and 55k isoforms. J Cell Physiol. 2005;203:251-60
35. Abudurexiti M, Zhu W, Wang Y, Wang J, Xu W, Huang Y. et al. Targeting CPT1B as a potential therapeutic strategy in castration-resistant and enzalutamide-resistant prostate cancer. Prostate. 2020;80:950-61
36. Hu Q, Poulose N, Girmay S, Heleva A, Doultsinos D, Gondane A. et al. Inhibition of CDK9 activity compromises global splicing in prostate cancer cells. RNA Biol. 2021;18:722-9
37. Rahman R, Rahaman MH, Hanson AR, Choo N, Xie J, Townley SL. et al. CDK9 inhibition inhibits multiple oncogenic transcriptional and epigenetic pathways in prostate cancer. Br J Cancer. 2024;131:1092-105
38. Fujinaga K, Huang F, Peterlin BM. P-TEFb: The master regulator of transcription elongation. Mol Cell. 2023;83:393-403
39. Wang Z, Yu H, Bao W, Qu M, Wang Y, Zhang L. et al. Proteomic and phosphoproteomic landscape of localized prostate cancer unveils distinct molecular subtypes and insights into precision therapeutics. Proc Natl Acad Sci U S A. 2024;121:e2402741121
40. Dong B, Xu JY, Huang Y, Guo J, Dong Q, Wang Y. et al. Integrative proteogenomic profiling of high-risk prostate cancer samples from Chinese patients indicates metabolic vulnerabilities and diagnostic biomarkers. Nat Cancer. 2024;5:1427-47
41. Hsin JP, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26:2119-37
42. Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O. et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2015;29:1437-41
43. Jatwani K, Sarangdhar M, Kolli V, Khosla AA, Gopalakrishnan D. Risk assessment of myelosuppression with PARP inhibitors in prostate cancer: Insights from FAERS. J Clin Oncol. 2024;42(Suppl 16):12144
44. Goncalves E, Ryan CJ, Adams DJ. Synthetic lethality in cancer drug discovery: challenges and opportunities. Nat Rev Drug Discov. 2026;25:22-38
45. Seed G, Beije N, Yuan W, Bertan C, Goodall J, Lundberg A. et al. Elucidating acquired PARP inhibitor resistance in advanced prostate cancer. Cancer Cell. 2024;42:2113-23 e4
46. Tsujino T, Takai T, Hinohara K, Gui F, Tsutsumi T, Bai X. et al. CRISPR screens reveal genetic determinants of PARP inhibitor sensitivity and resistance in prostate cancer. Nat Commun. 2023;14:252
47. Li L, Karanika S, Yang G, Wang J, Park S, Broom BM. et al. Androgen receptor inhibitor-induced "BRCAness" and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal. 2017;10:eaam7479
48. Ye Q, Ma B, Li L, Wang Z, Lu M, Kang J. et al. A Peptide-Based PROTAC Degrader of BRCA2 Sensitizes Metastatic Castration-Resistant Prostate Cancer to PARP Inhibition. Cancer Res. 2025;85:3490-502
49. Wu C, Peng S, Pilie PG, Geng C, Park S, Manyam GC. et al. PARP and CDK4/6 Inhibitor Combination Therapy Induces Apoptosis and Suppresses Neuroendocrine Differentiation in Prostate Cancer. Mol Cancer Ther. 2021;20:1680-91
50. Wang LY, Hung CL, Wang TC, Hsu HC, Kung HJ, Lin KH. PROTACs as Therapeutic Modalities for Drug Discovery in Castration-Resistant Prostate Cancer. Annu Rev Pharmacol Toxicol. 2025;65:375-96
51. Rahaman MH, Kumarasiri M, Mekonnen LB, Yu M, Diab S, Albrecht H. et al. Targeting CDK9: a promising therapeutic opportunity in prostate cancer. Endocr Relat Cancer. 2016;23:T211-T26
52. Paparidis NF, Durvale MC, Canduri F. The emerging picture of CDK9/P-TEFb: more than 20 years of advances since PITALRE. Mol Biosyst. 2017;13:246-76
53. Yu DS, Zhao R, Hsu EL, Cayer J, Ye F, Guo Y. et al. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010;11:876-82
54. Zou Y, Zhang H, Chen P, Tang J, Yang S, Nicot C. et al. Clinical approaches to overcome PARP inhibitor resistance. Mol Cancer. 2025;24:156
55. Chu YY, Chen MK, Wei Y, Lee HH, Xia W, Wang YN. et al. Targeting the ALK-CDK9-Tyr19 kinase cascade sensitizes ovarian and breast tumors to PARP inhibition via destabilization of the P-TEFb complex. Nat Cancer. 2022;3:1211-27
56. Hu C, Shen L, Zou F, Wu Y, Wang B, Wang A. et al. Predicting and overcoming resistance to CDK9 inhibitors for cancer therapy. Acta Pharm Sin B. 2023;13:3694-707
57. Toure MA, Motoyama K, Xiang Y, Urgiles J, Kabinger F, Koglin AS. et al. Targeted degradation of CDK9 potently disrupts the MYC-regulated network. Cell Chem Biol. 2025;32:542-55 e10
Author contact
![]() Corresponding authors: Jun Yan, Ph.D., Fudan University, Shanghai 200032, China. Phone: 86-21-54237454; E-mail: yan_junedu.cn. Ruimin Huang, Ph.D., Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. Phone: 86-21-68077978; E-mail: rmhuangac.cn. Rujian Zhu, M.D., Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, China. Phone: 86-18918753975; E-mail: tzzhurjcom.
Corresponding authors: Jun Yan, Ph.D., Fudan University, Shanghai 200032, China. Phone: 86-21-54237454; E-mail: yan_junedu.cn. Ruimin Huang, Ph.D., Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. Phone: 86-21-68077978; E-mail: rmhuangac.cn. Rujian Zhu, M.D., Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, China. Phone: 86-18918753975; E-mail: tzzhurjcom.