Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
1. Critical roles of Macrophages...
2. The Emergence of...
3. The Immunological Mechanisms...
4. Diagnosis and Prognosis of...
5. Conclusions and Perspectives
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2026; 16(13):7715-7734. doi:10.7150/thno.127500 This issue Cite
Review
Understanding immune checkpoint inhibitors-related myocarditis: Mechanisms and targeted therapeutic pathways
Wenxiao Xia, Huan Zhang, Libo Liu* ![]() , Chenchen Tu*
, Chenchen Tu* ![]() , Xiantao Song*
, Xiantao Song* ![]()
Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, China.
*These authors have contributed equally to this work.
Received 2025-10-29; Accepted 2026-3-12; Published 2026-6-17
Abstract

Immune checkpoint inhibitor (ICI)-related myocarditis is a rare but often fatal adverse event that has gained increasing attention due to the widespread clinical use of ICIs. This review explores the development of immune cells and their critical roles in cardiac autoimmunity, with particular focus on defects in central tolerance, the role of immune checkpoints, T cell trafficking to the heart, and the involvement of resident cardiac macrophages. It also introduces the key pathogenic mechanisms of ICI-related myocarditis, including the activation of autoreactive T cells that recognize self or shared antigens, the crosstalk between T cells and macrophages, and macrophage polarization. In addition, we also discuss current and emerging targetable therapeutic pathways, including cytokine modulation, the JAK/STAT pathway, and the use of CTLA-4 immunoglobulin. We further summarize practical diagnostic and prognostic approaches, including the role and limitations of troponin-based monitoring, echocardiography, magnetic resonance imaging, PET-CT applications, and endomyocardial biopsy. This review aims to establish a mechanistic framework that integrates pathogenic mechanisms, diagnostic and prognostic approaches, and therapeutically targetable pathways in ICI-related myocarditis.
Keywords: immune checkpoint inhibitors, myocarditis, cardiac autoimmunity, targeted therapies
Introduction
The development of immune checkpoint inhibitors (ICIs) has revolutionized cancer therapy, shifting the focus from conventional chemotherapy and radiotherapy to the promising realm of immunotherapy. By targeting key immune checkpoints, including cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed death-1 (PD-1), and programmed cell death 1 ligand 1 (PD-L1), ICIs reactivate T cell-mediated antitumor responses [1]. Since the approval of the first anti-CTLA-4 agent for the treatment of advanced melanoma in 2011, ICIs have demonstrated remarkable clinical efficacy and an acceptable safety profile, effectively halting tumor progression and extending patient survival while further expanding their use in cancer treatment. However, in clinical practice, ICIs may induce a series of side effects known as immune-related adverse events (irAEs). The occurrence of irAEs increases the complexity of the treatment and deserves further attention. Reported cases of irAEs frequently include dermatological toxicities, colitis, hepatitis, pneumonitis, nephritis, and a range of endocrine disorders [2]. Among these, the incidence of immune-related cardiac toxicity remains relatively low [3]; however, fulminant ICI-related myocarditis, with grave complications, including cardiogenic shock, heart failure, and life-threatening arrhythmias, exhibits a significantly higher mortality rate than other irAEs, ranging from 25% to 50% [4,5]. These adverse reactions highlight the balance between the therapeutic benefits of immunotherapy and its potential deleterious effects on cardiac function. In this review, we provide a spatiotemporal perspective on the intricate interplay between T cell development and cardiac autoimmunity. Subsequently, we discuss the distinct cellular compositions of cardiac macrophages and T lymphocytes, specifically highlighting their contributions to the maintenance of cardiac immune homeostasis. We further summarize practical diagnostic and prognostic approaches that combine biomarker surveillance with multimodal imaging in ICI-related myocarditis. This foundational context paves the way for a thorough exploration of the pathogenesis and targeted pathways for ICI-related myocarditis.
1. Critical roles of Macrophages and T Lymphocytes in Cardiac Autoimmunity
Healthy cardiac tissue harbors all major leukocyte populations, including monocytes/macrophages, neutrophils, B cells, and T cells [6,7]. Lymphocytes account for 21%, and myeloid cells for 74% of cardiac immune cells, with macrophages representing the predominant myeloid population [8]. However, in the steady state, the heart is not an immunologically active organ, and immune cells represent only a small fraction of cardiac cellularity. The distribution of these major cell types in the human heart has been explored using single-cell RNA sequencing (scRNA-seq), and differences between the atrial and ventricular tissues have been found[9]. Cardiomyocytes accounted for 30.1% of the atrial tissue, and immune cells (both myeloid and lymphoid) accounted for 10.4%. In contrast, the proportion of ventricular cardiomyocytes increased to 49.2%, whereas the proportion of immune cells decreased to 5.3% [9]. However, the heart is not entirely immune-privileged. Its dense vascular and lymphatic networks enable immune cell trafficking and antibody access, and play crucial roles in pathogen clearance, inflammatory responses, limiting excessive immune damage, and immune tolerance [10]. Here, we review the process of T cell development and its intersection with cardiac immunity, while also emphasizing the role of T cells and macrophages in cardiac immune homeostasis.
1.1 The Interplay of Spatiotemporal T Cell Development and Cardiac Immunity
1.1.1 The Paradox of T Cell Central Tolerance and Cardiac Autoimmunity
T lymphocytes play a role in adaptive immune responses, which are essential for forming defense against infections and sustaining immune homeostasis. However, they can also initiate hypersensitivity reactions and autoimmune diseases. T cells originate in the bone marrow and subsequently migrate to the thymus, where they undergo central tolerance. Within the thymic cortex, immature CD4⁺CD8⁺ double-positive (DP) T cells interact with peptide-loaded major histocompatibility complex (pMHC) molecules presented by cortical thymic epithelial cells (cTECs) [11] (Figure 1). DP T cells that fail to receive adequate T-cell receptor (TCR) signaling through these interactions undergo apoptosis by “death by neglect” within 3–4 days [12]. Surviving thymocytes are positively selected to enforce MHC restriction and subsequently commit to CD4⁺ or CD8⁺ single-positive (SP) T cells based on the preferential recognition of MHC class II or class I, respectively [13]. SP T cells then migrate into the medulla, where antigen-presenting cells (APCs) including medullary thymic epithelial cells (mTECs) and dendritic cells (DCs), implement negative selection to purge autoreactive TCR specificities [14]. Promiscuous gene expression in mTECs enables them to express a substantial repertoire of tissue-restricted antigens (TRAs), typically found exclusively in specific tissues or cell types [15]. Collectively, impairment of negative selection may allow autoreactive thymocytes to escape deletion and exit thymus, providing a mechanistic basis for autoimmune predisposition.
Cardiac immune homeostasis and the fate of cardiac autoreactive T lymphocytes. T cells undergo central tolerance in the thymus and then enter the periphery. Loss of peripheral tolerance confers upon them a migratory capacity, facilitating chemotactic recruitment to cardiac tissue and the initiation of an immune response. Specifically, double-positive thymocytes within the thymus, subjected to positive selection, differentiate into single-positive T cells. Given the absence within mTECs of genes encoding certain cardiac-specific tissue-restricted antigens (e.g., Myh6), potentially autoreactive T cells transit to the periphery. After exposure to cardiac-derived self-antigens and specific cardiac homing factors (e.g., HGF) following the failure of peripheral tolerance based on immune checkpoints for some reason, T cells acquire a cardiac trafficking label (e.g., c-Met+ CXCR3+ CCR4+) and infiltration into the myocardium, triggering an autoimmune inflammatory response in concert with macrophages.
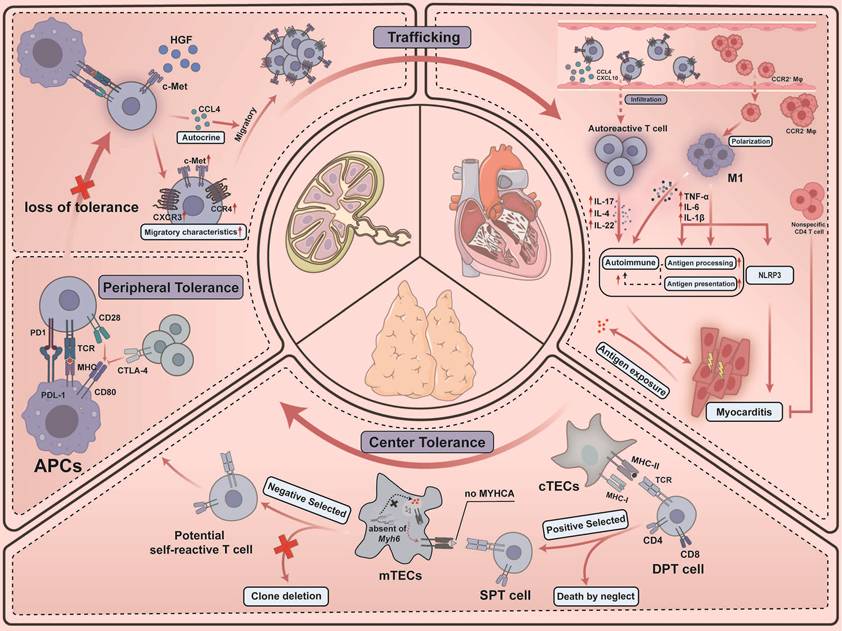
However, central tolerance exhibits an intrinsic vulnerability with respect to cardiac antigens, rendering the cardiac tissue particularly susceptible to autoimmune injury. Lv et al. discovered transcripts for Myh6 (encoding α-myosin heavy chain, MYHCA) were absent from murine and human mTECs [16]. Such distinct lapses in tolerance predispose the myocardium to autoimmunity, allowing latent populations of autoreactive T cells to persist in peripheral tissues until they are provoked by extrinsic stress. Specifically, the sterile inflammatory response to tissue necrosis mobilizes autoreactive T cells that evade thymic deletion. Borght et al. showed that myocardial infarction triggers cDC2 migration to draining lymph nodes, where presentation of MYHCA autoantigen promotes differentiation of quiescent CD4⁺ T cells into pathogenic Th1/Th17 effectors [17]. Subsequent work by Forte et al. further revealed that the cDC1 subset orchestrates antigen cross-presentation through CLEC9A, a receptor that helps process antigens from necrotic cells and cross-priming of cytotoxic CD8+ T cells, thereby awakening latent autoreactive immune potential [18]. Moreover, Cunha-Neto et al. identified CD4⁺ T-cell clones recognizing cardiac myosin in patients with Chagas disease and in corresponding animal models, consistent with a mechanism in which the Trypanosoma cruzi–derived B13 protein cross-reacts with MYHC epitopes and thereby activates latent MYHC-autoreactive CD4⁺ T cells [19]. Notably, even in the absence of tissue damage, MYHCA can be constitutively presented to CD4+ T cells by cardiac APCs via MHC class II [17], and T cells derived from CTLA-4-deficient mice undergo vigorous spontaneous proliferation and precipitate severe ICI-related myocarditis [20] (see Section 3.1).
Taken together, cardiac TRA deficiency within mTECs presents a paradox: central T cell tolerance coexists with susceptibility to cardiac autoimmunity. Cardiac autoimmunity is fundamentally rooted in the intrinsic limitations of central tolerance. Thymic escape of cardiac-reactive T cells establishes a latent immunological substrate, whereas ischemic or infectious insults act as catalysts that convert this vulnerability into an overt pathology. This paradigm is further corroborated by the observation that alterations in the thymic epithelium, particularly in thymic epithelial tumors, are associated with both the incidence and severity of ICI-related myocarditis [21].
1.1.2 T cell activation and trafficking to the heart
During the initiation of adaptive immune responses, naïve T cells encounter DCs within secondary lymphoid organs, where DCs present MHC molecules complexed with antigenic peptides [22]. If TCRs specifically recognize these antigenic peptides and T cells simultaneously receive co-stimulatory signals, for example, through the interaction of CD28 with B7 family molecules (CD80, CD86), T cells initiate a specific transcriptional program that drives the functional differentiation of effectors, thus eliciting an immune response [23]. CD4+ T cells release cytokines to attract and activate other immune cells, while CD8+ T cells gain cytotoxic abilities by directly eliminating the infected or neoplastic cells. Most effector T cells subsequently undergo apoptosis after pathogen clearance, while a subset persists as memory T cells [24]. The surviving memory T cells constitute the predominant circulating population in the blood and are further classified into distinct subsets based on their phenotypes, functions, and migratory patterns: central memory T cells (Tcm, CD45RA- CCR7+), effector memory T cells (Tem, CD45RA- CCR7-), and stem cell memory T cells (Tscm, CD45RA+ CCR7+ CD95+ CD122+) [25,26]. This classification method may be flawed, however, because there may be overlap between different subpopulations and cells within the same subpopulation may have different functional properties [27]. Moreover, although the majority of effector T cells succumb to apoptosis following infection, it has been observed in humans that a population of terminal effector cells displaying the CD45RA+ CCR7- phenotype, known as Temra cells, can persist within the circulation [28]. Temra cells represent a highly differentiated subset within the human CD8+ memory T cell lineage, and they increase with age [28]. While possessing a limited proliferative capacity, Temra cells can produce significant amounts of IFN-γ and IL-2, as well as express elevated levels of perforin and FasL, endowing them with potent cytotoxic capabilities [29].
Defining the determinants of organ-specific T cell trafficking, including the heart, remains challenging. Distinct memory T cell subsets exhibit organ-specific homing capabilities, interacting with the endothelial cells (ECs) of their respective target tissues, and soluble factors secreted by the tissues themselves may contribute to the directed recruitment of these T cell populations [30]. Specifically, within the lymph nodes (Figure 1), hepatocyte growth factor (HGF) derived from the heart binds to the c-Met receptor on T cells, thereby inducing memory T cells to acquire distinctive migratory characteristics (c-Met+ CCR4+ CXCR3+) that are specific to the heart [31]. Furthermore, c-Met signaling promotes the autonomous release of chemokines (e.g., CCL4), thereby establishing an autocrine chemokine loop that further enhances the migration of T cells toward the heart [31]. This increase in circulating c-Met+ memory T cells may delineate the characteristics of adaptive cardiac inflammation, and the elevation of autoantigen-specific c-Met+ T cells in peripheral blood marks the loss of cardiac immune tolerance [32]. In humans, c-Met+ T cells infiltrate inflamed myocardium in acute myocarditis and idiopathic dilated cardiomyopathy, respond to the cardiac myosin and secrete cytokines such as IL-4, IL-17, and IL-22 [32]. In addition, scRNA-seq analysis of cardiac CD45+ cells showed that cytotoxic T cells were the largest T cell population in the normal heart, and Th17 cells appeared in large numbers in the early stages of cardiac autoimmune acute inflammation and reached a peak at day 14 [33]. In later stages, myocardial non-specific CD4+ T cells were effective in inhibiting post-inflammatory fibrotic remodeling [34].
In effector T cell-mediated immune responses, antigen-specific activated lymphocytes must be efficiently localized to antigen-rich nonlymphoid tissues, and the HGF-c-Met signaling pathway defines the homing of memory T cells toward the heart [32]. The balance among T cell populations functions as a double-edged sword; however, the precise roles and mechanisms of various cell subsets remain a subject of debate [33,34]. Defining the molecular determinants of cardiac homing and resolving the functional heterogeneity among cardiac-infiltrating T cells remains a priority.
1.2 Diverse Macrophage Subsets in Cardiac Immune Homeostasis and Autoimmunity
Resident cardiac macrophages (RCMs) are the most abundant immune cells in the adult heart, accounting for 6% to 10% of non-cardiomyocytes [35]. Myocardium contains distinct RCM subsets classified by their divergent origins and marker expression [36]: Monocytes in the circulatory system can express CCR2, a chemokine receptor crucial for migration [37], and these monocytes can infiltrate locally into affected tissues, replenishing RCM populations and contributing to the progression of cardiovascular disease [38]. In contrast, CCR2- RCMs originate from embryonic progenitor cells in the yolk sac and maintain the mostly resident population of cardiac tissue at homeostasis by localized proliferation [39]. Upon myocardial injury, RCMs populations expand, in part because CCR2+ RCMs release MCPs to recruit additional monocytes via a MYD88-dependent mechanism, rather than enhancing ECs adhesion or transendothelial migration [40]. As immune profiling has advanced, additional macrophage subsets have been identified. Based on common life cycle properties and core gene signatures in mouse hearts, scRNA-seq identifies three distinct subsets of macrophages: TLF+ macrophages (TIMD4+ LYVE1+ FOLR2+ MHC-IIlo CCR2-) are sustained independent of blood monocytes; MHC-IIhi macrophages (TIMD4-LYVE1- FOLR2- MHC-IIhi CCR2-) are partially replaced by monocytes; and CCR2+ macrophage subsets (TIMD4-LYVE1-FOLR2- MHC-IIhi CCR2+) are entirely substituted by monocytes [41,42]. Across these lineages, tissue niches shape macrophage programs via cues such as IL-34 and M-CSF [43,44], consistent with expression of CD163, COLEC12, MRC1, MARCH1, and NRAMP1 [6].
Macrophages also dominate autoimmune myocarditis [33]. Infiltrating cardiac monocytes can differentiate into M1-type macrophages (Figure 1), which release pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, thereby precipitating tissue damage and an inflammatory cascade [45]. In experimental autoimmune myocarditis (EAM), acute inflammation features an inflammation-associated macrophage cluster enriched for Hif1a-related genes and nitric oxide production, supporting enhanced antigen processing and endogenous peptide presentation [33]. Hua et al. demonstrated that the STING inhibitor C-176 alleviates myocardial inflammation and functional impairment in EAM mice, affirming STING's role in enhancing pro-inflammatory macrophage polarization via the HIF1α pathway and thereby contributing to the pathogenesis of autoimmune myocarditis [46]. Furthermore, macrophages engage in the NLRP3 inflammasome-mediated pyroptosis that exacerbates EAM progression through the PPARα/LACC1/NF-κB signaling cascade [47].
In the preclinical ICI-related myocarditis model using Ctla4⁺/⁻Pdcd1⁻/⁻ mice, GO enrichment analysis indicates that CCR2⁻ CD163⁺ cardiac macrophages are preferentially engaged in regulating epithelial cell proliferation as well as in orchestrating stress responses [48]. This profile mirrors the role of CCR2⁻ RCMs as a homeostatic subpopulation essential for tissue maintenance and anti-inflammatory regulation [49]. Consistently, in endomyocardial biopsy specimens from patients with ICI-related myocarditis, CCR2⁻ macrophages defined by CD163, LYVE1 and FOLR2 expression retain homeostatic and tissue-support programs without evidence of inflammatory reprogramming [50]. By contrast, in Ctla4⁺/⁻Pdcd1⁻/⁻ mice, an IFN-γ–inducible Cxcl9⁺Cxcl10⁺ macrophage subset has been identified, characterized by chemokine production and enhanced antigen-presenting capacity [48]. Trajectory analysis, together with CCR2 blockade using the MC-21 antibody, indicates that these cells arise from CCR2⁺ monocyte-derived macrophages [48]. In ICI-related myocarditis, CCR2⁺ macrophages thus acquire IFN-γ–driven antigen-presenting and chemokine-producing programs and undergo selective expansion [51], establishing CCR2⁺ macrophages as a central pathogenic population that amplifies myocardial injury through the CXCL9/CXCL10–CXCR3 axis (see also Section 3.2.1).
2. The Emergence of Cardiotoxicity Associated with Immune Checkpoint Inhibitors
2.1 The Multifaceted Roles of Immune Checkpoints in Cardiac Autoimmunity
Examples of α-myosin–reactive T cells underscore that not all cardiac TRAs are represented in the thymus [16], permitting thymic escape of autoreactive clones. Peripheral tolerance therefore becomes essential, and immune checkpoints constitute a central inhibitory layer within this system. As inhibitory receptor–ligand pairs on immune cells, CTLA-4 and PD-1/PD-L1 function as core “brakes” that set activation thresholds for T cells and help preserve cardiac immune homeostasis.
2.1.1 CTLA-4
CTLA-4 and CD28 are homologous receptors expressed on both CD4+ and CD8+ T cells [52]. CTLA-4, with a higher affinity for the CD28 ligands CD80 and CD86, outcompetes CD28 for binding, thereby blocking co-stimulatory signaling and inducing T cell anergy, a state where T cells, despite recognizing an antigen, fail to become fully activated and proliferate [52]. Furthermore, CTLA-4 can physically limit the availability of costimulatory molecules. Emerging research indicates that CTLA-4 can engage in trans-endocytosis, which directly captures CD80 and CD86 from the surface of APCs and internalizes them for degradation [53]. This mechanism directly reduces the costimulatory capacity of APCs, providing an additional layer of immune regulation [54]. However, the trans-endocytosis model requires further investigation to fully elucidate its intricacies and implications.
The immunomodulatory role of CTLA-4 in the heart is similar to its role in other tissues. It inhibits T-cell activation and proliferation, thereby maintaining homeostasis and reducing autoimmune attacks in cardiac tissues [55]. Congenital CTLA-4-deficient mice develop fatal myocarditis, whereas adult-deficient mice do not have significant, death-inducing cardiac pathologic alterations, suggesting that the role of CTLA-4 in cardiac immune tolerance may be related to the developmental period [56]. Furthermore, CTLA-4 plays a key role in the maintenance of allograft tolerance by reducing rejection of the transplanted heart and affecting long-term graft survival by inhibiting T-cell activation [57].
2.1.2 PD-1/PD-L1
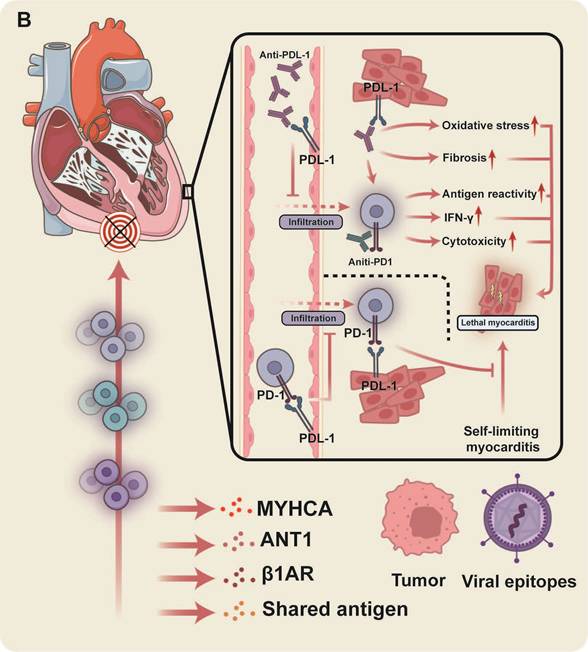
PD-1 is an inhibitory receptor expressed on the surface of T cells that engages with its ligands, PD-L1 and PD-L2, which are primarily presented by APCs and other cell types. PD-L1 is a ubiquitous ligand that is widely expressed across multiple cells, including T cells, B cells, APCs, vascular and stromal endothelial cells, and pancreatic islet cells [58,59], whereas the expression of PD-L2, a ligand predominantly expressed by DCs, macrophages, and B cells, is more restricted [58,59]. Unlike CTLA-4, PD-1 primarily suppresses T cell activation through signal transduction pathways. Mechanistically, PD-1 inhibits Akt phosphorylation by blocking CD28-mediated PI3K activation, thereby downregulating transcriptional programs responsible for T cell activation, proliferation, and survival [60]. During the effector phase of the immune response, the absence of PD-1 expression can lead to an increase in T cell proliferation or specific effector functions [61]. Moreover, PD-1 deficiency also skews CD8+ T cells towards a Tcm phenotype, characterized by elevated expression levels of CD62L, CD27, and CCR7, and enhanced IL-2 production [62].
PD-L1 is expressed in the heart, and markedly elevated levels of PD-L1 mRNA have been detected in mouse cardiac tissue [63]. PD-L1 expression induced on cardiac ECs plays a critical immunomodulatory role (Figure 2B). Endothelial cells activated by IFN-γ inhibit T cell activation through the expression of PD-L1 and PD-L2, and blocking PD-L1 with antibodies enhances the costimulatory effects of endothelial cells on CD8+ T cells, amplifying their responsiveness to antigen presentation by ECs, which includes increased IFN-γ secretion and augmented cytotoxic activity [64]. Consistent with this, studies involving genetically modified mice lacking PD-L1 and PD-L2 have demonstrated that the induced expression of PD-L1 on cardiac ECs plays a crucial role in immune modulation within the heart. This mechanism can safeguard cardiac tissue from damage inflicted by CD8+ T cells and the inflammation induced by polymorphonuclear leukocyte-rich microabscesses, thus preventing otherwise self-limiting myocarditis from progressing into a life-threatening condition [65]. Moreover, the inflammation and oxidative stress resulting from PD-1 deficiency may precipitate electrical and structural remodeling of the atria, thereby increasing the risk of atrial fibrillation [66]. In neonatal mice, PD-1 deficiency exacerbates the inflammatory response following cardiac injury, as evidenced by increased monocyte infiltration, heightened expression of pro-inflammatory factors, and worsened fibrosis [67]. Concurrently, DN T cells in these mice display heightened cytotoxic sensitivity to cardiac antigens, underscoring the critical role of the PD-1/PD-L1 pathway in cardiac protection and regeneration by inhibiting inflammation and autoimmune damage [67]. Although PD-L1 has been extensively studied in the context of cardiac immune homeostasis, the specific role of PD-L2 remains to be elucidated. In EAM models, PD-L2 deficiency is associated with exacerbated myocardial inflammation, and flow cytometric analyses have revealed a significant increase in inflammatory cells [68]. Moreover, DCs lacking PD-L2 exhibited enhanced proliferation of CD4+ T cells in response to TCR and CD28 signaling, suggesting that PD-L2 may play a critical role in curbing the expansion of autoreactive CD4+ T cells and subsequent inflammation [68].
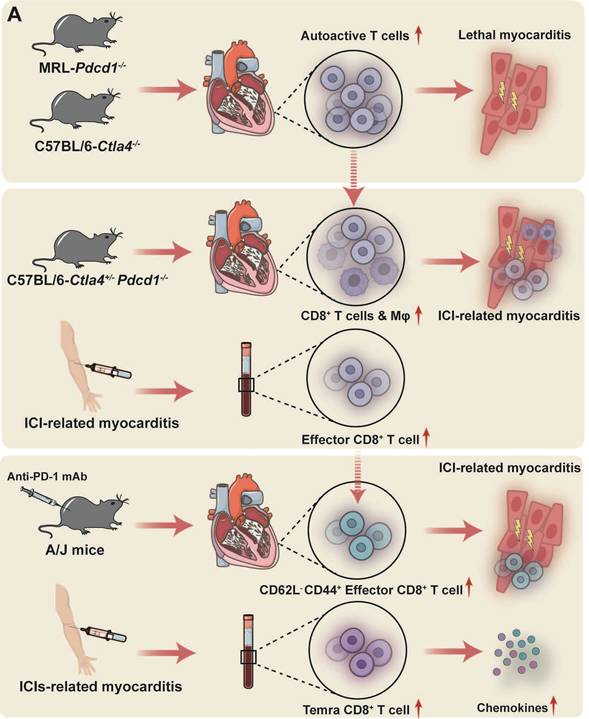
The primary pathogenic mechanisms underlying ICI-related myocarditis. (A) Exacerbated autoreactive CD8+ T cell activity is a key driver in ICI-associated myocarditis and insight into the precise pathogenic T cell subsets is progressively clarifying. (B) MYHCA is a primary target antigen for cardiac T cell attack, though additional autoantigens, or antigens shared by tumors and viruses, potentially contribute. The presented figure demonstrates a protective role for PD-1/PD-L1 within the heart and the detrimental consequences of disrupting this axis.
2.2 The Application of Immune Checkpoint Inhibitors and Related Myocarditis
Immune checkpoints are essential for maintaining immune homeostasis and preventing immune system hyperactivity by promoting peripheral tolerance mechanisms. However, tumor cells can exploit these checkpoint mechanisms to evade immune surveillance, thereby enhancing their survival and proliferation [69]. Specifically, tumor cells induce immune resistance and T cell exhaustion by expressing checkpoint proteins such as PD-L1, which inhibit T cell activation and proliferation through the engagement of these inhibitory molecules [69]. Given that these checkpoints can be readily blocked by antibodies or regulated through the reconfiguration of ligands and receptors [70], ICIs targeting proteins, such as CTLA-4, PD-1, and PD-L1 have been developed to relieve the immune suppression and reactivate T cell-mediated immune responses, thereby inducing attacks against tumors [1].
ICIs therapy has fundamentally transformed the field of oncology. Since the approval of the first anti-CTLA-4 agent for advanced melanoma in 2011, ICIs have heralded a new era of cancer treatment. Subsequently, various ICIs have been approved for the treatment of melanoma, lung cancer, colorectal cancer, hepatocellular carcinoma, and renal cell carcinoma have been approved for clinical use, offering patients novel therapeutic alternatives [71-74]. These ICIs include anti-CTLA-4 monoclonal antibodies (mAbs) such as ipilimumab, anti-PD-1 mAbs (pembrolizumab, nivolumab, and cemiplimab), anti-PD-L1 mAbs (atezolizumab, avelumab, and durvalumab), and anti-LAG-3 mAbs. Collectively, they exhibit remarkable clinical efficacy and an acceptable safety profile, effectively inhibiting tumor progression and prolonging survival, while their indications continue to expand.
Despite their efficacy, ICI-induced immune activation can lead to a range of side effects known as irAEs. In addition to issues related to drug resistance that limit the number of patients who can achieve sustained benefits [75], the occurrence of irAEs adds complexity to treatment and warrants increasing attention. In cancer patients receiving ICIs, reported cases of irAEs commonly include skin toxicity, colitis, hepatitis, pneumonitis, nephritis, and endocrine disorders, such as thyroid dysfunction [2].
Recently, cardiac toxicity has emerged as a major concern. Although immune-related cardiac toxicity is less frequent than other irAEs, the estimated one-year risk of developing pericarditis or myocarditis is 1.8% [3]. However, due to its potential to lead to severe outcomes such as cardiogenic shock, heart failure, and fatal arrhythmias, the associated mortality rate is significantly higher, ranging from 25% to 50% compared to other irAEs [4,5]. Additionally, an analysis of the burden of cardiac disease toxicity associated with ICIs in cancer patients aged 65-85 years indicates that cardiac disease is one of the primary disease types resulting from these ICIs [76]. This underscores the importance of early recognition and management of immune-related cardiac toxicity. Pharmacovigilance analysis of ICIs has identified potential ICI-related major cardiac adverse drug reactions, including myocarditis, atrial fibrillation, heart failure, pericardial effusion, tachycardia, myocardial infarction, sinus tachycardia, acute myocardial infarction, pericarditis, and cardiac tamponade [5,77]. Among fatal ICI-related cardiac adverse drug reactions, myocarditis is the most common, accounting for 50.8% of cases, making it a crucial focus in ICI cardiotoxicity research [5].
2.3 Temporal Characteristics of Myocarditis Onset
The onset of myocarditis after ICIs therapy exhibits significant individual variability. Most patients develop symptoms during the early stages of treatment, with the earliest manifestations potentially occurring after the first administration of ICI therapy. Specifically, Mahmood et al. conducted a retrospective case-control study and found that the median time to the onset of myocarditis after the initiation of ICIs therapy was 34 days, with 81% of cases occurring within three months of treatment initiation [4]. Similarly, a pharmacovigilance study of cases in the VigiBase database reported a median time to onset of 33 days (interquartile range: 21–91 days) of myocarditis symptoms after the first administration of ICIs therapy [77]. However, a small cohort of patients, presents with delayed-onset ICI-related myocarditis, which manifests several months or even longer after the commencement of treatment. In a notable report by Yamaguchi et al., a case of late-onset fulminant myocarditis was observed after 13 cycles of nivolumab therapy [78]. Furthermore, delayed immune-related events following ICI discontinuation are gaining recognition. Nguyen et al. reported the longest latency period yet for ICI-related myocarditis, with a 33-week interval between pembrolizumab cessation for metastatic colorectal cancer and myocarditis diagnosis, exceeding two years from the initiation of treatment [79]. These findings underscore the surveillance of immune-related cardiac toxicity. The diagnosis of ICI-related myocarditis and meticulous monitoring of the cardiac status should not be confined solely to the early phases of ICI treatment; rather, vigilance must be maintained, even during prolonged therapy and after the cessation of treatment.
2.4 Multidrug Combinations as Principal Risk Factors
Monotherapy with PD-1/PD-L1 inhibitors has been linked to an increased risk of cardiovascular adverse events (CVAEs) across all severity grades[80]. Notably, treatment groups receiving anti-PD-1 antibodies demonstrate a higher incidence of cardiac adverse events (69.4%) compared to those treated with anti-CTLA-4 antibodies (20%) [5]. Among all therapeutic regimens, combined PD-L1 and CTLA-4 blockade carries the highest risk of CVAEs of any grade [81]. The analysis of the VigiBase pharmacovigilance database corroborates this observation, indicating that combination ICI therapy is associated with a higher mortality rate for myocarditis compared to ICI monotherapy [77].
Klocke et al. demonstrated that thymic escape of CTLA-4–deficient autoreactive clonal T cells establishes a foundational immunological substrate for myocarditis susceptibility [56]. In parallel, Wei et al. revealed a gene dosage–dependent functional interplay between CTLA-4 and PD-1, whereby Ctla4 haploinsufficiency combined with Pdcd1 loss precipitates fulminant ICI-related myocarditis [82]. Furthermore, genetic or pharmacologic dual checkpoint disruption accelerates CD8⁺ T-cell and CCR2⁺ macrophage myocardial infiltration and activates the CXCL9/CXCL10–CXCR3 axis, establishing a feed-forward loop in myocarditis [51]. Collectively, CTLA-4 and PD-1 operate at complementary checkpoints of the T-cell response, with CTLA-4 constraining priming and costimulation in lymphoid organs and PD-1 limiting effector activity in peripheral tissues, including the heart. Beyond emphasizing the need for intensified baseline counseling and early surveillance with dual regimens, these mechanistic insights provide a rationale for restoring co-inhibitory signaling using CTLA-4-Ig in steroid-refractory ICI-related myocarditis (see Section 3.5).
3. The Immunological Mechanisms and Potential Therapeutic Targets in ICI-Related Myocarditis
3.1 The Excessive Activation of Autoreactive T Cells is Central to the Pathogenesis of ICI-related Myocarditis
3.1.1 Autoreactive CD8+ T Cells
Preclinical models and human data converge on the critical role of T cells in the pathogenesis of ICI-related myocarditis, highlighting the overactivation of autoreactive T cells as a necessary condition for its development. The MRL-Pdcd1-/- mouse model spontaneously develops lethal myocarditis, characterized by a massive infiltration of lymphocytes and myeloid cells within cardiac tissue [83]. Adoptive transfer experiments further support the notion that PD-1 deficiency leads to the proliferation and activation of autoreactive T cells [83]. Mice deficient in Ctla4 exhibit a phenotype analogous to that of Pdcd1-deficient mice, albeit with greater severity, characterized by persistent T cell activation, infiltration across multiple organs, and subsequent tissue damage, ultimately leading to premature mortality [20].
Wei and colleagues established a C57BL/6 background model featuring Ctla4+/- and Pdcd1-/- strains, whereby premature mortality was attributed to cardiac T cell and macrophage infiltration, alongside significant electrocardiographic abnormalities, mirroring the clinical and pathological features of ICI-related myocarditis observed in patients [82]. Notably, this study validated that the gene dosage of Ctla4 and Pdcd1 collectively dictates the threshold for T cell self-recognition, with their functional interaction deemed critical for maintaining cardiovascular homeostasis [82].
Further characterization of the T cell phenotype revealed a substantial presence of highly activated and proliferating, clonal CD8+ T cells within the myocardium of Pdcd1-/-Ctla4+/- mice [82,84]. Adoptive transfer of immune cells from myocarditis mice into Rag1-/- recipients induced lethal myocarditis; conversely, CD8+ T cells depletion abrogated this induction [84]. Consistent with these findings, scRNA-seq of peripheral immune cells from patients with PD-1 inhibitor-related myocarditis revealed intense peripheral immune responses, specifically an increased proportion of CD8+ T cells [85]. Analogous findings have been observed in an inducible myocarditis pharmacological model. Administration of anti-PD-1 mAb to A/J mice successfully induced ICI-related myocarditis, resulting in robust activation of autoreactive CD8+ T cells [86]. Specifically, a significant elevation in the proportion of CD62L-CD44+ effector T cell subsets was observed within the myocardium of the affected mice, whereas the proportion of CD62L+CD44+ memory T cell subsets remained comparable to controls [86]. A comprehensive analysis of peripheral blood mononuclear cells (PBMCs) from patients with ICI-related myocarditis, utilizing CyTOF, scRNA-seq, and TCR-seq, revealed a significant expansion of Temra CD8+ T cells in the peripheral blood [87]. These Temra CD8+ T cells exhibit pronounced activation and cytotoxic potential, while the frequencies of other major circulating immune populations were largely unchanged [87]. Notably, these cells demonstrate elevated expression of chemokines CCL5, CCL4, and CCL4L2, potentially promoting cardiac T cells migration and contributing to the pathogenesis of ICI-related myocarditis [87]. Recent investigations have implicated a protective role for CD4+ TCM cells in ICI-related myocarditis injury, and their proportion is inversely correlated with myocarditis severity, which stems from the elevated expression of immunosuppressive factors such as IL-4I1, effectively mitigating the proliferation and activation of Temra CD8+ T cells [88]. Further studies are needed to elucidate the precise mechanisms underlying the protective function of CD4+ TCM cells in ICI-related myocarditis.
3.1.2 Cardiac Self-antigens and Shared Antigens as a Key to T Cell-mediated Attacks on the Heart
What initiates the transition from autoreactive potential to overt myocardial injury remains an active question. The prevailing hypotheses posit that cardiac-specific proteins, particularly MYHCA, are pivotal in driving effector T cells toward an autoimmune assault on the heart in patients with ICI-related myocarditis. Previous sections have delineated the inherent vulnerability of cardiac immune tolerance mechanisms, specifically, the insufficient expression of MYHCA in mTECs. This deficiency prevents the deletion of α-myosin-reactive T cells within the thymus, allowing their release into the peripheral circulation. In Pdcd1-/-Ctla4+/- mice, analysis of the TCR repertoire identified α-myosin as the target antigen for three dominant clonal TCRs in this model of ICI-related myocarditis [84]. Further investigation revealed the presence of α-myosin-specific TCRs in blood samples from ICI-related myocarditis patients, alongside significant clonal expansion of these TCRs in both the cardiac and skeletal muscles of these patients [84]. Moreover, the treatment of A/J mice with anti-PD-1 antibodies may reactivate previously quiescent MYHCA-specific T cells and potentially precipitate myocarditis [86]. Recent findings by Kalinoski et al. demonstrated the presence of autoreactive MYHCA tissue resident memory (TRM) cells co-expressing PD-1 and CD69 within the healthy heart; during post-injury cardiac recovery stages, these TRM cells accumulate within the epicardium, which may increase susceptibility to ICI-related cardiomyopathy [89].
Beyond MYHCA, additional cardiac self-antigens have been proposed. Employing A/J mice bearing distinct adenine nucleotide translocase 1 (ANT1) peptide sequences, Basavalingappa et al. demonstrated that the mitochondrial protein ANT1 can induce EAM, with the ANT1 21-40 peptide fragment identified as the principal myocarditis epitope, and associated with IL-17A production by T cells [90]. Immunization of A/J mice with specific β1AR peptide sequences revealed their ability to bind major histocompatibility complex class II/IAk or IEk alleles, thereby inducing varying degrees of myocarditis [91]. This β1AR peptide-mediated response further demonstrated an enhanced clonal expansion of TCRs [92].
Tumor cells may express antigens homologous or identical to those found in normal tissues, potentially leading to autoimmune toxicity against healthy tissues during the immune system's recognition and attack of tumor cells. This cross-reactivity, which induces autoreactive T cells, has been implicated in lung irAEs [93]. Case reports have documented the frequent presence of shared T cell receptor sequences in cardiac, skeletal muscle, and tumor tissues. The elevated expression of muscle-specific transcripts within tumor tissues further supports the possibility of cross-reactivity with autoantigens, including Myh6 [94]. Analysis of the TCGA melanoma cohort reveals detectable Myh6 expression in 69% of cases [84]. However, approximately 40% of melanoma tumor cells aberrantly express Myh6 in ICI-treated patients without clinically significant myocarditis or myositis [84]. A case study of three patients developing severe myocarditis and myositis within three weeks of both COVID-19 booster vaccination and PD-1 blockade therapy revealed overlapping T cell receptor repertoires, potentially due to cross-reactivity between pre-existing T cell responses to viral epitopes (e.g., the spike protein) elicited by mRNA COVID-19 vaccines and muscle antigens [95]. Although cross-reactivity between tumor and autoantigens does not appear to be a determinant factor in ICI-related myocarditis, further investigations are necessary to uncover potential yet-unrecognized cross-reactive pathways.
In summary, excessive activation of autoreactive T cells serves as a critical prerequisite for ICI-related myocarditis. Both animal models and human data indicate that deficiencies in PD-1 or CTLA-4 lead to the proliferation and activation of autoreactive T cells, ultimately culminating in cardiac inflammation and tissue damage (Figure 2A). Cardiac-specific proteins, particularly MYHCA, are considered key elicitors of T cell-mediated attacks on the heart, although contributions from other self-antigens and the possibility of epitope spreading from tumors should also be considered (Figure 2B).
3.2 Macrophages: an Indispensable Component
3.2.1 The CXCL9/CXCL10-CXCR3 Axis
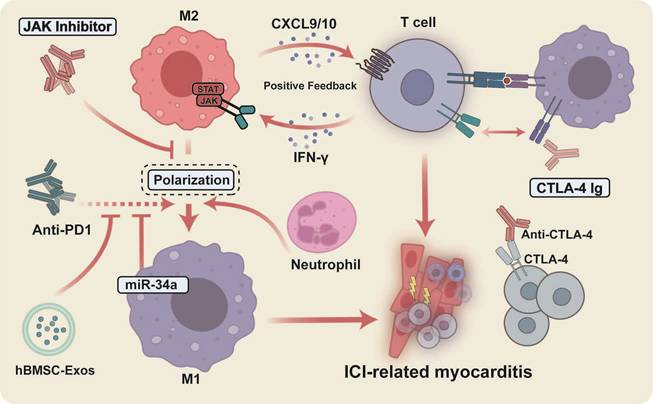
Given the characteristic macrophage infiltration observed in ICI-related myocarditis, the interplay between T cells and macrophages has received heightened scrutiny in understanding the pathogenesis of ICI-related myocarditis. In the Ctla4+/-Pdcd1-/- mice, Pan et al. identified a CXCL9+CXCL10+ CCR2+ macrophage subset within cardiac tissue [48]. These macrophages are derived from circulating CCR2+ monocytes and are shaped by T cell-derived IFN-γ; a similar macrophage subpopulation has also been observed in human cases of ICI-related myocarditis [48]. CD8+ T cells depletion or the blockade of the IFN-γ signaling pathway resulted in a reduction of CXCL9+CXCL10+ macrophages and an amelioration of myocarditis symptoms in murine models [48]. Huang et al. demonstrated the accumulation of CXCL9/10+ CCR2+ macrophages and clonally expanded CXCR3hiCD8+ T cells in the MRL-Pdcd1-/- model [51]. By selectively targeting and disrupting the interactions between these T cells and the CXCL9/10 released by macrophages, the research prevented and treated ICI-related myocarditis in the model [51]. This finding further supports the notion that the IFN-γ signaling pathway, along with the CXCL9/CXCL10-CXCR3 axis, mediates a positive feedback loop between T cells and macrophages, thereby exacerbating cardiac inflammation and injury.
3.2.2 Macrophage Polarization Intensifies Myocarditis
PD-1 inhibitors are associated with M1 polarization of cardiac macrophages and increased expression of pro-inflammatory mediators, including iNOS, IL-1β, IL-6, and TNF-α, consistent with amplified inflammatory responses and myocardial injury [96]. Mechanistically, TargetScan analysis and luciferase assays demonstrate that miR-34a targets the 3' untranslated region of KLF4 [96]. Inhibition of miR-34a or overexpression of KLF4 effectively reverses PD-1 inhibitor-induced M1 polarization and cardiac damage, suggesting that KLF4 exerts a protective anti-inflammatory effect, whereas miR-34a promotes cardiac injury by suppressing KLF4 in the heart. [96]. In addition, macrophages can affect the cell cycle and telomere length through the accumulation of miR-34a-5p in exosomes and its translocation into cardiomyocytes, which in turn targets the 3'-untranslated region of the serine/threonine protein phosphatase 1 regulatory subunit 10 (PNUTS) of cardiomyocytes, leading to cardiac senescence and dysfunction [97]. These findings provide novel insights into the mechanisms underlying ICI-related cardiac injury, underscoring the need for further investigations to substantiate the relationship between miR-34a and immunotherapy-induced cardiotoxicity.
Antibodies targeting CTLA-4 m2a may exacerbate cardiac injury in EAM mice by promoting the infiltration of neutrophils, particularly a subset of Ccl5-expressing neutrophils, which in turn drives macrophages toward a pro-inflammatory M1 phenotype, resulting in overall cardiac inflammation, fibrosis, and dysfunction [98]. Extracellular vesicles derived from human bone marrow mesenchymal stem cells (hBMSC-Exos) can mitigate cardiac damage in melanoma mice induced by BMS-1 (PD-1/PD-L1 inhibitor). These hBMSC-Exos inhibit the M1 macrophage polarization induced by BMS-1 while promoting a shift toward M2 macrophage polarization, accompanied by reduced expression levels of pyroptosis-related proteins such as ASC, caspase-1, NLRP3, and GSDMD [99].
In summary, by modulating the interplay between CD8+ T cells and macrophages (Figure 3), as well as the polarization of macrophages—particularly through the inhibition of M1 macrophage polarization, it is possible to effectively attenuate the inflammatory response and damage associated with myocarditis, thereby offering novel insights and potential therapeutic targets for the treatment of this condition.
Therapeutic interventions for ICI-related myocarditis focus on modulating the interplay between CD8+ T cells and macrophages, specifically M1 polarization. Positive feedback loops driven by the IFN-γ/JAK/STAT1 and CXCL9/10/CXCR3 axes, promoting T cell–macrophage synergy and M1 macrophage polarization, amplify immune-mediated inflammation and tissue damage in ICI-related myocarditis. JAK inhibition represents a promising novel therapeutic avenue targeting this mechanism.
3.3 Exploratory Cytokine Targeting Pathways for ICI-related Myocarditis
Building on the pathogenic framework outlined above, in which autoreactive T cells and inflammatory macrophages form a self-reinforcing circuit, cytokines can be viewed as network-level amplifiers that couple cellular crosstalk to tissue injury. In ICI-related myocarditis models, macrophage-derived cues (e.g., IL-6 and IL-23) promote effector skewing and expansion of pathogenic CD4⁺ T-cell states [100,101], whereas T-cell–derived cytokines (notably IL-17A) act downstream to potentiate myocardial inflammation [102], remodeling and functional decline. Importantly, these mediators are not introduced here as isolated biomarkers but represent intervenable targets that mechanistically connect the previously described T-cell–macrophage interactions with organ-level cardiotoxicity, while offering a rational opportunity to decouple antitumor efficacy from ICI-related toxicity.
In a preclinical pharmacological model of PD-1 blockade, CD4⁺ T cells are skewed towards Th17 differentiation, thereby emerging as the predominant source of IL-17A [103]. This shift is mirrored by increased Th17 infiltration of the myocardium, coupled with upregulated Il17a and RORγt expression and enhanced IL-17A staining within cardiac tissue [103]. Consistently, in two cases of ICI-related myocarditis, IL-17 staining of endomyocardial biopsy specimens suggested myocardial Th17 infiltration [104]. In the TnI–induced autoimmune myocarditis (TnI-AM) mouse model, Bockstahler et al. showed that cytokines from CD11b⁺ monocyte–derived cells, particularly IL-6, not only amplify inflammation but also expand autoreactive CD4⁺ T cells and bias their differentiation toward a Th17 phenotype, which is associated with more severe cardiac inflammation and fibrosis [104]. Genetic deletion of the immunoproteasome component LMP7 or pharmacological inhibition with ONX 0914 markedly reduced cardiac IL-17 expression and alleviated myocardial inflammation [104]. Importantly, the TnI-AM model does not recapitulate ICI-related myocarditis. In contrast, in murine models of ICI-related myocarditis, the upstream cues that commit CD4⁺ T cells to Th17 remain incompletely defined. What is clearer from available intervention studies is that Th17-derived IL-17A acts as a key downstream inflammatory mediator of ICI-related cardiotoxicity, aligning with adverse cardiac remodeling, fibrosis, and a heart failure–like functional decline [102]. In PD-1 blockade models, heightened IL-17 signaling accompanies cardiac dysfunction, whereas IL-17A neutralization (or CD4⁺ T-cell depletion) can prevent deterioration in cardiac performance [102].
IL-6 is a crucial cytokine in the differentiation of naïve CD4+ T cells into Th17 cells, a process that is intricately linked to the pathogenesis of various autoimmune diseases. In addition to the aforementioned findings in the TnI-AM mouse model [104], IL-6 released by CD11b⁺ macrophages drives CD4⁺ T cell differentiation towards a Th17 phenotype, which is closely associated with exacerbated cardiac inflammation and fibrosis [105]. Chen et al. established a lung cancer mouse model of PD-1 inhibitor–induced myocarditis and, by dual immunofluorescence staining, identified infiltrating M1 macrophages as a major source of cardiac IL-6 [101]; elevated IL-6 in turn activates the cardiomyocyte JAK2/STAT3 axis, drives oxidative stress, and reinforces a feed-forward loop of M1 polarization and cytokine release [101]. Because STAT3 is also a well-recognized pro-tumor effector downstream of IL-6 [106], IL-6R blockade with tocilizumab is mechanistically attractive: it may preferentially restrain the IL-6–Th17 axis to dampen inflammation while largely preserving Th1 function and CD8⁺ cytotoxic activity, and simultaneously attenuate JAK2/STAT3-dependent tumor growth and metastatic signaling [101]. Retrospective studies found that treating irAEs with anti-IL-6R therapy in melanoma patients increased the objective response rate from 56% to 68%, although evidence for the resolution of ICI-related myocarditis and its translation into clinical benefit remains limited [107].
In numerous autoimmune diseases, IL-23 drives the excessive proliferation and activation of inflammatory cells, thereby directly contributing to inflammation and tissue damage while playing a pivotal role in the underlying pathological processes of the disease [108]. In the cohort of patients with irAEs, IL-23 levels increased with irAE severity and exhibited a monocyte-dominant expression profile in peripheral blood mononuclear cell transcriptomes, indicating upstream signaling driven by myeloid cells [100]. Consistent with this, prophylactic IL-23 blockade effectively prevented myocarditis induced by dual CTLA-4 and PD-1 immunotherapy in humanized mouse models without compromising the antitumor efficacy of the combined immunotherapy [100]. This suggests that IL-23 may serve as an intervenable node for decoupling therapeutic efficacy from toxicity. It is noteworthy that in ICI-related colitis, myeloid-derived IL-23 synergizes with the CXCL9/10–CXCR3 axis to expand Th17 cells and drive their shift towards a pathogenic IFN-γ⁺ IL-17⁺ dual-function state [109]. This mechanism bears a resemblance to the CXCL9/CXCL10–CXCR3 positive feedback amplification circuit observed in ICI-related myocarditis in terms of its network architecture.
In summary, cytokine-targeted strategies offer a mechanistically grounded approach to disrupting the T cell–macrophage–driven inflammatory amplification loop in ICI-related myocarditis. However, the causal hierarchy and dominant amplification nodes among individual cytokines remain incompletely defined. Future studies integrating single-cell communication and spatial omics, particularly in models that better recapitulate the tumor–immune–cardiac microenvironment, will be essential. Clinically, a key challenge is whether cytokine blockade can mitigate cardiotoxicity without compromising antitumor efficacy, particularly in patients receiving combination ICI regimens. Addressing this trade-off will require prospective studies with paired cardiac endpoints and oncologic outcomes, rather than treating cardiac safety as an isolated objective.
3.4 The Inhibition of the IFN-γ-JAK-STAT1 Pathway
IFN-γ secreted by CD8+ T cells exacerbates ICI-related myocarditis through crosstalk with Cxcl9+Cxcl10+ macrophages, and the underlying mechanism has been described above. The findings from mouse and human studies support the potentially beneficial role of JAK inhibitors in ICI-related myocarditis. Specifically, Ctla4+/-Pdcd1-/- mice exhibited significantly enhanced STAT1 expression and phosphorylation levels, while the use of the JAK1/2 inhibitor, ruxolitinib, significantly inhibited IFN-γ-induced Cxcl9 expression in BMDMs in vitro, suggesting that IFN-γ-JAK-STAT1 signaling plays an important role in the emergence of Cxcl9+Cxcl10+ macrophages [48]. Bulk RNA-seq from ICI-related myocarditis patients also showed that JAK2 was the only gene in the JAK family that was significantly upregulated in ICI-related myocarditis patients compared to ICI-treated patients without evidence of myocarditis [110]. There is growing evidence for the clinical efficacy of JAK inhibitors in a variety of autoimmune and inflammatory diseases, including autoimmune rheumatic diseases and inflammatory bowel diseases [111,112]. Previous studies demonstrated that JAK inhibitors exhibit potential efficacy in the treatment of refractory ICI-associated colitis [113,114]. Case reports have indicated that personalized JAK inhibitor therapy offers additional clinical benefit in corticosteroid-refractory ICI-related myocarditis, providing a novel option for intensified immunosuppression [115,116] (Table 1). Consistent with this, a recent multicenter observational study involving 24 patients with corticosteroid-resistant ICI-related myocarditis demonstrated a slight to moderate decline in cTnT levels after treatment with tofacitinib, a JAK1/3 inhibitor [117]. However, among the five patients with life-threatening fulminant myocarditis who were unresponsive to both corticosteroids and tofacitinib, no sustained decrease in cTnT levels was observed [117]. This indicates that tofacitinib may demonstrate differential efficacy across various forms of ICI-related myocarditis, particularly as an adjunctive therapy for patients who are either resistant to or have failed to successfully taper the corticosteroid treatment. However, in the critical group, tofacitinib alone may not be sufficient to control the disease, which may be related to the fact that tofacitinib primarily inhibits JAK1/3, rather than JAK2. However, tofacitinib demonstrated a favorable safety profile in terms of toxicity and antitumor activity in cancer patients who developed irAEs [117]. Furthermore, in a cohort of 30 patients with ICI-related myocarditis who underwent a modified treatment regimen, the incorporation of systematic respiratory muscle screening and mechanical ventilation, along with high-dose abatacept and ruxolitinib therapy, significantly reduced the mortality associated with myotoxicity from 60% to 3.4% [110].
References for targeted treatment strategies in patients with ICI-related myocarditis
| References (Year) | Study type | Number of patients treated | Targeted treatment strategy | Outcome evaluation | Conclusion |
|---|---|---|---|---|---|
| Salem, et al.[110] (2023) | Single center, prospective, observational cohort study | 30 patients treated with the modified strategy | Systematic screening and management of respiratory muscle involvement + mechanical ventilation when needed + high-dose abatacept + ruxolitinib + corticosteroids | Myotoxicity-related fatality decreased from 60% in the initial guideline-treated cohort to 3.4% (1/30) after implementation of the modified strategy. | Early recognition and management of respiratory muscle involvement, combined with CD86 receptor occupancy-guided abatacept and ruxolitinib, may reduce the high fatality rate of severe ICI-related myocarditis. |
| Liu, et al.[117] (2024) | Multicenter, retrospective, observational study | 53 irAE patients, including 48 with myocarditis | Tofacitinib-based immunosuppression, mostly 5 mg twice daily, with or without additional immunosuppressive therapy | Clinical remission rates were 54.5%, 96.7%, and 100% in the life-threatening, steroid-resistant, and steroid taper-failure groups, respectively. | Tofacitinib showed promising efficacy in irAEs, particularly in patients with steroid resistance or steroid taper failure; however, its efficacy may be limited in life-threatening fulminant myocarditis. |
| Xing, et al.[116] (2022) | Case Report | 1 | Tofacitinib | Patient recovered clinically during the hospitalization without major adverse cardiac events. | Tofacitinib can offer additional clinical benefit in steroid-refractory cases and provides a new option for intensified immunosuppressive therapy in ICI-related myocarditis. |
| Nguyen, et al.[115] (2022) | Case Report | 1 | Personalized-dose-adjusted abatacept + ruxolitinib + corticosteroids | Clinical improvement occurred within 7 days, with resolution of systolic cardiac dysfunction and ventricular arrhythmias, followed by successful discharge. | Personalized dose adapted immunosuppression using abatacept, ruxolitinib, and corticosteroids achieved quick reversal of fulminant, life-threatening ICI-related myocarditis refractory to standard therapy. |
| Salem, et al.[119] (2019) | Case Report | 1 | Abatacept | Cardiac troponin T levels decline rapidly and ventricular arrhythmias resolve within 3 weeks. | Abatacept may be effective in the treatment of severe ICI-related myocarditis refractory to glucocorticoid therapy. |
| Liu, et al.[120] (2020) | Case Report | 1 | Abatacept + mycophenolate mofetil + corticosteroids ± plasmapheresis | Heart failure symptoms improved, BNP decreased, and hsTnI stabilized and gradually decreased but remained persistently elevated. | Although aggressive multidrug combination immunosuppressive therapy (including Abatacept and MMF) may improve clinical symptoms, it may not completely resolve myocardial injury (as evidenced by persistent troponin elevation) |
Overall, these preliminary studies suggest that JAK inhibitors may have potential benefits in the treatment of ICI-related myocarditis. However, further investigation into the mechanisms of the JAK pathway in this context, along with larger-scale prospective studies, is essential to confirm their efficacy and safety.
3.5 CTLA-4 Ig as a Promising Therapeutic Approach
Genetic studies have shown that Ctla4 and Pdcd1 interact in a gene dose-dependent manner to determine the frequency and severity of ICI-related myocarditis, while epidemiological evidence has also identified combined ICI therapy as a risk factor for this condition.
Abatacept, a CTLA-4-Fc fusion protein, interacts with the CD80/CD86 molecules on the surface of antigen-presenting cells, such as macrophages, effectively blocking the co-stimulatory signaling pathways essential for T cell activation [82,118]. This mechanism differs from that of broad-spectrum immunosuppressants, potentially enabling a more targeted approach to mitigate ICI-related myocarditis, thereby reducing adverse effects. In the Ctla4+/- Pdcd1-/- murine model, treatment with abatacept resulted in a notable decrease in mortality and an improvement in cardiac pathology [82]. Preliminary clinical evidence in the reported case series supports the use of CTLA-4 Ig for the treatment of ICI-related myocarditis, while abatacept may be an effective therapeutic option for severe and corticosteroid-refractory ICI-related myocarditis [82,119,120] (Table 1). Concerns have been raised regarding the potential of CTLA-4 Ig, owing to its inhibitory effects on co-stimulation, to compromise the associated antitumor responses elicited by ICIs.
However, recent investigations have demonstrated that the co-administration of CTLA-4 Ig with ICIs may diminish antitumor efficacy, whereas its application after ICI therapy can enhance therapeutic outcomes [118]. Post-immunotherapy treatment with CTLA-4 Ig does not significantly modify the expression of co-stimulatory molecules on APCs or influence the frequency or cytokine production of CD8+ T cells, suggesting that once CD8+ T cells achieve full activation following ICI therapy, continued co-stimulation may be less essential and does not hinder antitumor efficacy; rather, post-treatment CTLA-4 Ig enhances the therapeutic effect through a critical mechanism that involves reducing the frequency of Tregs [118]. Moreover, Liu et al. evaluated belatacept and M17-2, which can bind to B7-1/B7-2 while failing to engage the clinically used anti-CTLA-4 antibodies, and these soluble CTLA-4 variants demonstrate superior efficacy in alleviating the severity of ICI-related myocarditis in murine models compared to abatacept, while concurrently preserving the antitumor efficacy of immunotherapy [121].
In summary, herein, we discussed the critical roles of autoreactive T cells, macrophages, and cytokines in the pathogenesis of ICI-related myocarditis. Hyperactivation of CD8+ T cells, especially in the absence of PD-1 and CTLA-4, leads to infiltration of autoreactive CD8+ T cells and macrophages, which triggers severe cardiac inflammation. Notably, the CXCL9/CXCL10-CXCR3 axis mediates interactions between T cells and macrophages, which exacerbate myocardial injury. Novel therapeutically targetable pathways, including targeting cytokines, JAK inhibitors, and CTLA-4, have shown potential to control myocarditis without significantly compromising antitumor efficacy. Consistent exploration of the potential mechanisms and emerging therapeutic options is essential to advance the treatment of ICI-related myocarditis and optimize patient prognosis.
4. Diagnosis and Prognosis of ICI-related Myocarditis
ICI-related myocarditis is characterized by an unpredictable time to onset and disproportionately high mortality rate, underscoring the critical need for early diagnosis and accurate risk stratification. Given the limited mechanistic clarity and the lack of targeted therapies, a proactive surveillance strategy that integrates biomarkers and multimodal imaging is essential for early detection and prognostic assessment.
4.1 Endomyocardial Biopsy (EMB)
EMB remains the histopathological gold standard for myocarditis diagnosis; however, its clinical utility in ICI-related myocarditis is constrained by sampling error, procedural risk, and the limited sensitivity of the Dallas criteria [122]. ICI-related myocarditis is characterized by prominent CD68⁺ macrophage infiltration with abundant CD4⁺ and CD8⁺ T cells and myocardial PD-L1 upregulation [123]. The incorporation of immunohistochemistry (CD3, CD8, and CD68) substantially improves diagnostic sensitivity compared with H&E staining alone [124]. Moreover, pathological grading based on the inflammatory burden correlates with serum troponin levels and clinical outcomes, providing a potential prognostic value [125,126]. Nevertheless, EMB is not recommended as a first-line diagnostic modality because of safety concerns and lack of evidence supporting its routine use [127].
4.2 Cardiovascular Magnetic Resonance (CMR)
CMR has emerged as the most informative noninvasive imaging modality for both the diagnosis and risk stratification of ICI-related myocarditis. In 2018, the updated version of the LLC was published, revising the diagnostic criteria for acute myocarditis to require the presence of at least one T2-based marker indicative of myocardial edema, and at least one T1-related marker reflecting myocardial damage (such as abnormal T1, extracellular volume, or LGE), in order to confirm the diagnosis of AM [128,129]. Compared with other cardiac imaging modalities, CMR has a remarkable capacity for myocardial tissue characterization, rendering it a promising technique for identifying patients at high risk of ICI-related myocarditis.
A multicenter retrospective study encompassing 136 cases of ICI-related myocarditis revealed that 78% of patients exhibited abnormal T1 values, whereas 43% displayed altered T2 values [130]. Thavendiranathan et al. employed the updated Lake Louise criteria and discovered that nearly all patients with ICI-related myocarditis met the diagnostic standards for non-ischemic myocardial injury [130]. The LGE pattern observed in ICI-related myocarditis typically manifests as non-ischemic characteristics, predominantly localized to the mid-myocardial or epicardial regions, although it may also be present in the subendocardial layer or throughout the entire myocardial thickness [123]. The most frequently affected areas include the anteroseptal, inferoseptal, inferior wall, and inferolateral wall [123]. In over half of the patients with ICI-related myocarditis, the LVEF remains normal [131]. Nonetheless, LGE remains a crucial marker for diagnosing ICI-related myocarditis via CMR imaging, although its sensitivity may be reduced when LVEF is normal and CMR is performed early [131].
4.3 Echocardiography
Echocardiography is the first-line imaging modality because of its accessibility and feasibility. While reductions in LVEF are uncommon, myocardial strain parameters, particularly global longitudinal strain and basal segment involvement, are frequently impaired and strongly associated with elevated troponin levels and major adverse cardiac events [132-134]. Nonetheless, echocardiographic findings remain nonspecific and should be interpreted as supportive rather than diagnostic.
4.4 Positron Emission Tomography (PET)
Apart from suspected cases of cardiac sarcoidosis, 18F-fluorodeoxyglucose (FDG) is not routinely used in the clinical assessment of patients with myocarditis [135]. Similarly, current international guidelines do not recommend PET for the diagnostic evaluation of ICI-related myocarditis [136]. As a viable alternative for assessing myocardial inflammation, FDG-PET directly quantifies increased metabolic activity within the myocardial wall resulting from localized inflammatory cell activation [137]. This approach provides complementary clinical insights alongside CMR findings indicative of myocardial edema in the context of diagnosing ICI-related myocarditis [138]. Case reports have documented the ability of PET-CT to identify early-stage ICI-related myocarditis, particularly when CMR remains inconclusive [139-141]. Furthermore, a retrospective, single-center study comparing CMR and 68Ga-DOTATOC PET/CT revealed the high sensitivity (100%) of the latter for detecting ICI-related myocarditis, especially during the early inflammatory stages where clinical symptoms are apparent; however, CMR remains unremarkable [142]. PET also has the potential to detect concomitant myositis, thereby enhancing the comprehensive assessment of the disease extent.
4.5 Cardiac Troponin
Elevated troponin levels are a universal feature of ICI-related myocarditis [4,143,144]. A retrospective cohort study indicated that all individuals diagnosed with ICI-related myocarditis presented with abnormal high-sensitivity troponin T (hsTnT) levels, suggesting that low hsTnT levels may exclude the diagnosis of ICI-related myocarditis [143]. Nevertheless, the elevation of hsTnT levels warrants careful consideration, as it may be attributable to preexisting conditions or the direct cardiovascular effects of the underlying malignancy, rather than being a consequence of the ICI therapy itself [145]. In this context, baseline troponin measurements before ICI treatment initiation are of significant value. Furthermore, analysis of 30 patients' troponin T (hs-TnT) levels before ICI treatment revealed that those with baseline hs-TnT levels ≥14 ng/L exhibited a heightened risk of cardiovascular events or progressive cardiac involvement within three months [146]. Similarly, a retrospective analysis of 135 patients treated with pembrolizumab demonstrated that persistently elevated hs-TnI levels (>50 ng/L) at baseline and during follow-up were robust predictors of MACE, with baseline hs-TnI levels exhibiting a particularly strong correlation with all-cause mortality [147]. Troponin I is the preferred biomarker for ICI-related myocarditis, because it more rapidly reflects myocardial injury in conditions such as myocarditis and exhibits greater specificity than creatine kinase (CK) and troponin T [148,149]. However, subsequent research has indicated that cTnT may exhibit superior efficacy in assessing the risk of MACE. When cTnT levels exceeded 1.5 ng/ml, the risk of MACE escalated fourfold (HR 4.0; 95% CI 1.5 to 10.9; p = 0.003) [144]. Similarly, in a prospective cohort study encompassing 60 patients with confirmed ICI-related myocarditis followed for one year, cTnT exhibited superior diagnostic sensitivity (98% vs. 88% for cTnI within 72 hours of admission) and a heightened predictive capacity for MACE within 90 days, with a cTnT upper reference limit ≥ 32 times identified as the potential predictive threshold [150]. The optimal cardiac troponin thresholds for a clinically meaningful diagnosis and risk stratification remain elusive. Ischemic heart disease, akin to fulminant myocarditis, elicits pronounced elevation in cardiac troponin levels, thus necessitating the exclusion of acute coronary syndromes from differential diagnosis.
4.6 Non-cardiac Biomarkers
Furthermore, an observational cohort study revealed that patients with myocarditis frequently exhibit elevated levels of noncardiac biomarkers during treatment with ICI treatment, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and creatine phosphokinase (CPK) [143]. A multivariable analysis conducted by Alexi Vasbinder et al. identified that only CPK levels were significantly associated with the development of myocarditis (HR: 1.83; 95% CI: 1.59–2.10) and overall mortality (HR: 1.10; 95% CI: 1.01–1.20) [143]. Notably, elevated CPK levels demonstrated a sensitivity of 99% and specificity of 23% in the identification of myocarditis [143].
5. Conclusions and Perspectives
In this review, we provide a comprehensive review of ICI-related myocarditis, describing the role of the heart in immune homeostasis and elucidating the complex interplay between autoreactive T cells, macrophages, and cytokines in the pathogenesis of this disease. Specifically, the development of ICI-related myocarditis is rooted in the intrinsic vulnerability of cardiac immune tolerance. Incomplete thymic presentation of cardiac TRAs allows autoreactive T cells to persist, and immune checkpoint blockade lowers the activation threshold of these clones, enabling their expansion, myocardial infiltration, and immune-mediated injury [16,82,84]. In parallel, IFN-γ–driven inflammatory macrophage programs amplify CXCR3⁺ T-cell recruitment and activation, establishing a self-reinforcing macrophage and T cell inflammatory circuit that sustains myocardial inflammation and accelerates myocardial dysfunction [48,51]. Despite increasing mechanistic clarity, several questions remain unresolved and should define the next phase of investigation. In particular, the developmental origin of pathogenic cardiac CD8⁺ T cells requires definitive elucidation. Dominant clonotypes may derive from naïve T cells primed within secondary lymphoid organs, from the expansion and functional reprogramming of pre-existing cardiac TRM cells, or from context-dependent contributions of both pathways [89]. Moreover, the macrophage and T–cell amplification loop provides a framework for targeted immunomodulation: attenuating IFN-γ–JAK–STAT1 signaling or disrupting CXCL9/CXCL10–CXCR3 coupling may restrain upstream inflammatory propagation [48,51,110], whereas cytokine-directed strategies such as IL-17A or IL-23 blockade may reduce downstream tissue injury programs and, potentially, enable efficacy–toxicity decoupling [100,102]. Critically, therapeutic development must maintain antitumor efficacy as its central constraint. These imperatives underscore the need for paired oncologic endpoints and rigorous consideration of treatment timing, dosing, and patient selection, rather than evaluating cardiac outcomes alone [100,117,118]. Interpretation of mechanistic studies also requires caution. Many preclinical evidences are derived from mouse models, which are indispensable for pathway dissection and interventional testing [86,98,100,101], but cannot fully recapitulate the human immune environment or oncologic context. Accordingly, insights generated from experimental models should be validated in well-characterized patient cohorts, with confirmation in human tissue and clear links to clinically relevant outcomes. Looking forward, multi-omics strategies are positioned to resolve these gaps. Single-cell profiling has already revealed clinically relevant immune-state shifts in peripheral blood [85,151], while imaging and biomarkers remain central for diagnosis and risk stratification [130,144,150]. Integrating single-cell RNA sequencing with spatially resolved assays in endomyocardial biopsies, complemented by single-cell TCR profiling, should connect clonal expansion to anatomical localization and in situ immune circuits [48,50]. In parallel, blood multi-omics coupled with imaging phenotypes may help identify less invasive markers that predict onset, severity, and recurrence [123,143,150,151]. Finally, given the diagnostic challenges and disproportionately high mortality associated with ICI–related myocarditis, multicenter cohort studies across diverse regions will be essential to delineate disease heterogeneity and to improve the generalizability of risk stratification frameworks and therapeutic pathways.
In conclusion, while ICIs have revolutionized cancer therapy, we must carefully navigate the challenges encountered along this path. As we strive to elucidate the mechanisms by which ICIs exacerbate cardiac inflammation, the development of targeted therapies that alleviate these adverse effects, without sacrificing the potent antitumor responses elicited by immunotherapy, remains the central focus of current research and clinical practice.
Abbreviations
ICIs: immune checkpoint inhibitors
CTLA-4: Cytotoxic T-lymphocyte antigen 4
PD-1: Programmed death-1
PD-L1: Programmed cell death 1 ligand 1
irAEs: immune-related adverse events
scRNA-seq: Single-cell RNA sequencing
DP T cells: double positive T cells
pMHC: peptide-loaded major histocompatibility complex
cTECs: Cortical thymic epithelial cells
SP T cells: single-positive T cells
mTECs: medullary thymic epithelial cells
TRAs: tissue-restricted antigens
Myh6: the gene encoding α-myosin heavy chain
MYHCA: α-myosin heavy chain
PIA: post-infarction Autoimmunity
DCs: dendritic cells
TCR: T-cell receptor
Tcm: central memory T cells
Tem: effector memory T cells
Tscm: stem cell memory T cells
HGF: hepatocyte growth factor
EAM: experimental autoimmune myocarditis
RCMs: Resident cardiac macrophages
mAbs: monoclonal antibodies
PBMCs: peripheral blood mononuclear cells
CyTOF: time-of-flight mass cytometry
TRM: tissue resident memory
ANT1: adenine nucleotide translocase 1
KLF4: Krüppel-like factor 4
PNUTS: protein phosphatase 1 regulatory subunit 10
hBMSC-Exos: human bone marrow mesenchymal stem cells
Acknowledgements
Funding
This study was supported by Beijing Hospitals Authority “sailing” Program (YGLX202520), Beijing Hospitals Authority’s Ascent Plan (DFL20220603), and High-level public health technical talent construction project of Beijing Municipal Health Commission (Leading Talent-02-01).
Authorship contributions statement
Wenxiao Xia contributed to writing—original draft and visualization. Huan Zhang and Libo Liu contributed to writing—review & editing. Chenchen Tu contributed to conceptualization and writing—review & editing. Xiantao Song contributed to conceptualization, writing—review & editing, and supervision. All authors reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lee JY, Lee HT, Shin W, Chae J, Choi J, Kim SH. et al. Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat Commun. 2016;7:13354
2. Dolladille C, Ederhy S, Sassier M, Cautela J, Thuny F, Cohen AA. et al. Immune Checkpoint Inhibitor Rechallenge After Immune-Related Adverse Events in Patients With Cancer. JAMA Oncol. 2020;6:865
3. D’Souza M, Nielsen D, Svane IM, Iversen K, Rasmussen PV, Madelaire C. et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: a nationwide Danish study. Eur Heart J. 2021;42:1621-31
4. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM. et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J Am Coll Cardiol. 2018;71:1755-64
5. Rubio-Infante N, Ramírez-Flores YA, Castillo EC, Lozano O, García-Rivas G, Torre-Amione G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: a meta-analysis. Eur J Heart Fail. 2021;23:1739-47
6. Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC. et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142:466-82
7. Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA. et al. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018;22:600-10
8. Yu Y-RA, O’Koren EG, Hotten DF, Kan MJ, Kopin D, Nelson ER. et al. A Protocol for the Comprehensive Flow Cytometric Analysis of Immune Cells in Normal and Inflamed Murine Non-Lymphoid Tissues. PLOS ONE. 2016;11:e0150606
9. Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL. et al. Cells of the adult human heart. Nature. 2020;588:466-72
10. Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dubé KN. et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522:62-7
11. Takahama Y, Nitta T, Mat Ripen A, Nitta S, Murata S, Tanaka K. Role of thymic cortex-specific self-peptides in positive selection of T cells. Semin Immunol. 2010;22:287-93
12. Holländer GA, Krenger W, Blazar BR, Holländer GA, Krenger W, Blazar BR. Emerging strategies to boost thymic function. Curr Opin Pharmacol. 2010;10:443-53
13. Zerrahn J, Held W, Raulet DH. The MHC Reactivity of the T Cell Repertoire Prior to Positive and Negative Selection. Cell. 1997;88:627-36
14. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol. 2014;14:377-91
15. Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME. et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 2014;24:1918-31
16. Lv H, Havari E, Pinto S, Gottumukkala RVSRK, Cornivelli L, Raddassi K. et al. Impaired thymic tolerance to α-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121:1561-73
17. Van der Borght K, Scott CL, Nindl V, Bouché A, Martens L, Sichien D. et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017;18:3005-17
18. Forte E, Perkins B, Sintou A, Kalkat HS, Papanikolaou A, Jenkins C. et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology After Ischemic Tissue Damage in the Heart. Circulation. 2021;143:821-36
19. Cunha-Neto E, Teixeira PC, Fonseca SG, Bilate AM, Kalil J. Myocardial gene and protein expression profiles after autoimmune injury in Chagas’ disease cardiomyopathy. Autoimmun Rev. 2011;10:163-5
20. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541-7
21. Fenioux C, Abbar B, Boussouar S, Bretagne M, Power JR, Moslehi JJ. et al. Thymus alterations and susceptibility to immune checkpoint inhibitor myocarditis. Nat Med. 2023;29:3100-10
22. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309-20
23. ElTanbouly MA, Noelle RJ. Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat Rev Immunol. 2021;21:257-67
24. Künzli M, Masopust D. CD4+ T cell memory. Nat Immunol. 2023;24:903-14
25. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708-12
26. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B. et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. 2018;19:183-91
27. Jameson SC, Masopust D. Understanding Subset Diversity in T Cell Memory. Immunity. 2018;48:214-26
28. Poon MML, Caron DP, Wang Z, Wells SB, Chen D, Meng W. et al. Tissue adaptation and clonal segregation of human memory T cells in barrier sites. Nat Immunol. 2023;24:309-19
29. Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260-6
30. McCully ML, Ladell K, Hakobyan S, Mansel RE, Price DA, Moser B. Epidermis instructs skin homing receptor expression in human T cells. Blood. 2012;120:4591-8
31. Komarowska I, Coe D, Wang G, Haas R, Mauro C, Kishore M. et al. Hepatocyte Growth Factor Receptor c-Met Instructs T Cell Cardiotropism and Promotes T Cell Migration to the Heart via Autocrine Chemokine Release. Immunity. 2015;42:1087-99
32. Fanti S, Stephenson E, Rocha-Vieira E, Protonotarios A, Kanoni S, Shahaj E. et al. Circulating c-Met-Expressing Memory T Cells Define Cardiac Autoimmunity. Circulation. 2022;146:1930-45
33. Hua X, Hu G, Hu Q, Chang Y, Hu Y, Gao L. et al. Single-Cell RNA Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation. 2020;142:384-400
34. Zarak-Crnkovic M, Kania G, Jaźwa-Kusior A, Czepiel M, Wijnen WJ, Czyż J. et al. Heart non-specific effector CD4+ T cells protect from postinflammatory fibrosis and cardiac dysfunction in experimental autoimmune myocarditis. Basic Res Cardiol. 2020;115:6
35. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R. et al. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118:400-9
36. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B. et al. Embryonic and Adult-Derived Resident Cardiac Macrophages Are Maintained through Distinct Mechanisms at Steady State and during Inflammation. Immunity. 2014;40:91-104
37. Leuschner F, Courties G, Dutta P, Mortensen LJ, Gorbatov R, Sena B. et al. Silencing of CCR2 in myocarditis. Eur Heart J. 2015;36:1478-88
38. Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M. et al. CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci. 2018;3:230-44
39. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24:1234-45
40. Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I. et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res. 2019;124:263-78
41. Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S. et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol. 2022;7:eabf7777
42. Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29-39
43. Muñoz-Garcia J, Cochonneau D, Télétchéa S, Moranton E, Lanoe D, Brion R. et al. The twin cytokines interleukin-34 and CSF-1: masterful conductors of macrophage homeostasis. Theranostics. 2021;11:1568-93
44. Guilliams M, Thierry GR, Bonnardel J, Bajenoff M. Establishment and Maintenance of the Macrophage Niche. Immunity. 2020;52:434-51
45. Lafuse WP, Wozniak DJ, Rajaram MVS. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells. 2020;10:51
46. Hua X, Bao M, Mo H, Sun Z, Xu M, Chen X. et al. STING regulates the transformation of the proinflammatory macrophage phenotype by HIF1A into autoimmune myocarditis. Int Immunopharmacol. 2023;121:110523
47. Xiong Y, Zhang Z, Liu S, Shen L, Zheng L, Ding L. et al. Lupeol alleviates autoimmune myocarditis by suppressing macrophage pyroptosis and polarization via PPARα/LACC1/NF-κB signaling pathway. Phytomedicine. 2024;123:155193
48. Ma P, Liu J, Qin J, Lai L, Heo GS, Luehmann H. et al. Expansion of Pathogenic Cardiac Macrophages in Immune Checkpoint Inhibitor Myocarditis. Circulation. 2024;149:48-66
49. Yang S, Penna V, Lavine KJ. Functional diversity of cardiac macrophages in health and disease. Nat Rev Cardiol. 2025;22:431-42
50. Sun B, Xun Z, Zhou Z, Zhang N, Piao M, Li C. et al. Single-cell transcriptomic analysis deciphers the inflammatory microenvironment characterized by CXCL9+ fibroblasts and ACKR1+ endothelial cells in immune-related myocarditis. J Transl Med. 2025;23:555
51. Huang YV, Sun Y, Chou H, Wagner N, Vitale MR, Bayer AL. et al. Novel Therapeutic Approach Targeting CXCR3 to Treat Immunotherapy Myocarditis. Circ Res. 2025;136:473-90
52. Engelhardt JJ, Sullivan TJ, Allison JP. CTLA-4 Overexpression Inhibits T Cell Responses through a CD28-B7-Dependent Mechanism. J Immunol. 2006;177:1052-61
53. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM. et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science. 2011;332:600-3
54. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci. 2021;118:e2023739118
55. Love VA, Grabie N, Duramad P, Stavrakis G, Sharpe A, Lichtman A. CTLA-4 Ablation and Interleukin-12-Driven Differentiation Synergistically Augment Cardiac Pathogenicity of Cytotoxic T Lymphocytes. Circ Res. 2007;101:248-57
56. Klocke K, Sakaguchi S, Holmdahl R, Wing K. Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci. 2016;113:E2383-2392
57. Chandraker A, Huurman V, Hallett K, Yuan X, Tector AJ, Park C-H. et al. CTLA-4 Is Important in Maintaining Long-Term Survival of Cardiac Allografts. Transplantation. 2005;79:897-903
58. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. 2018;18:153-67
59. Burke KP, Chaudhri A, Freeman GJ, Sharpe AH. The B7:CD28 family and friends: Unraveling coinhibitory interactions. Immunity. 2024;57:223-44
60. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV. et al. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol Cell Biol. 2005;25:9543-53
61. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212:1125-37
62. Allie SR, Zhang W, Fuse S, Usherwood EJ. Programmed Death 1 Regulates Development of Central Memory CD8 T Cells after Acute Viral Infection. J Immunol. 2011;186:6280-6
63. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H. et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J Exp Med. 2000;192:1027-34
64. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T. et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117-26
65. Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ. et al. Endothelial Programmed Death-1 Ligand 1 (PD-L1) Regulates CD8+ T-Cell-Mediated Injury in the Heart. Circulation. 2007;116:2062-71
66. Fu G, Cao Y, Lu J, Li J, Liu L, Wang H. et al. Programmed cell death-1 deficiency results in atrial remodeling in C57BL/6 mice. Int J Mol Med. 2013;31:423-9
67. Vargas Aguilar S, Cui M, Tan W, Sanchez-Ortiz E, Bassel-Duby R, Liu N. et al. The PD-1-PD-L1 pathway maintains an immunosuppressive environment essential for neonatal heart regeneration. Nat Cardiovasc Res. 2024;3:389-402
68. Li S, Tajiri K, Murakoshi N, Xu D, Yonebayashi S, Okabe Y. et al. Programmed Death-Ligand 2 Deficiency Exacerbates Experimental Autoimmune Myocarditis in Mice. Int J Mol Sci. 2021;22:1426
69. Wolchok JD, Chan TA. Antitumour immunity gets a boost. Nature. 2014;515:496-8
70. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-64
71. Tykodi SS, Gordan LN, Alter RS, Arrowsmith E, Harrison MR, Percent I. et al. Safety and efficacy of nivolumab plus ipilimumab in patients with advanced non-clear cell renal cell carcinoma: results from the phase 3b/4 CheckMate 920 trial. J Immunother Cancer. 2022;10:e003844
72. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. 2010;363:711-23
73. Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. The Lancet. 2016;387:1540-50
74. Boukouris AE, Theochari M, Stefanou D, Papalambros A, Felekouras E, Gogas H. et al. Latest evidence on immune checkpoint inhibitors in metastatic colorectal cancer: A 2022 update. Crit Rev Oncol Hematol. 2022;173:103663
75. Bagchi S, Yuan R, Engleman EG. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol Mech Dis. 2021;16:223-49
76. Yang F, Shay C, Abousaud M, Tang C, Li Y, Qin Z. et al. Patterns of toxicity burden for FDA-approved immune checkpoint inhibitors in the United States. J Exp Clin Cancer Res. 2023;42:4
77. Gougis P, Jochum F, Abbar B, Dumas E, Bihan K, Lebrun-Vignes B. et al. Clinical spectrum and evolution of immune-checkpoint inhibitors toxicities over a decade-a worldwide perspective. eClinicalMedicine. 2024;70:102536
78. Yamaguchi S, Morimoto R, Okumura T, Yamashita Y, Haga T, Kuwayama T. et al. Late-Onset Fulminant Myocarditis With Immune Checkpoint Inhibitor Nivolumab. Can J Cardiol. 2018;34:812.e1-812.e3
79. Nguyen AT, Berry GJ, Witteles RM, Le DT, Wu SM, Fisher GA. et al. Late-Onset Immunotherapy-Induced Myocarditis 2 Years After Checkpoint Inhibitor Initiation. JACC CardioOncology. 2022;4:727-30
80. Liu S, Gao W, Ning Y, Zou X, Zhang W, Zeng L. et al. Cardiovascular Toxicity With PD-1/PD-L1 Inhibitors in Cancer Patients: A Systematic Review and Meta-Analysis. Front Immunol. 2022;13:908173
81. Liu M, Cheng X, Ni R, Zheng B, Huang S, Yang J. Cardiotoxicity of immune checkpoint inhibitors: A frequency network meta-analysis. Front Immunol. 2022;13:1006860
82. Wei SC, Meijers WC, Axelrod ML, Anang N-AAS, Screever EM, Wescott EC. et al. A Genetic Mouse Model Recapitulates Immune Checkpoint Inhibitor-Associated Myocarditis and Supports a Mechanism-Based Therapeutic Intervention. Cancer Discov. 2021;11:614-25
83. Wang J, Okazaki I, Yoshida T, Chikuma S, Kato Y, Nakaki F. et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol. 2010;22:443-52
84. Axelrod ML, Meijers WC, Screever EM, Qin J, Carroll MG, Sun X. et al. T cells specific for α-myosin drive immunotherapy-related myocarditis. Nature. 2022;611:818-26
85. Gao J, Wang Y, Lu L, Ma M, Ling J, Sun L. et al. Peripheral immune mapping and multi-omics analysis in Pd-1 inhibitor-induced myocarditis. J Leukoc Biol. 2023;114:164-79
86. Won T, Kalinoski HM, Wood MK, Hughes DM, Jaime CM, Delgado P. et al. Cardiac myosin-specific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. 2022;41:111611
87. Zhu H, Galdos FX, Lee D, Waliany S, Huang YV, Ryan J. et al. Identification of Pathogenic Immune Cell Subsets Associated With Checkpoint Inhibitor-Induced Myocarditis. Circulation. 2022;146:316-35
88. Yu J, Long B, Li Z, Tian X, Li D, Long J. et al. Central memory CD4+ T cells play a protective role against immune checkpoint inhibitor-associated myocarditis. Cardiovasc Res. 2024;120:1442-55
89. Kalinoski H, Daoud A, Rusinkevich V, Jurčová I, Talor MV, Welsh RA. et al. Injury-induced myosin-specific tissue-resident memory T cells drive immune checkpoint inhibitor myocarditis. Proc Natl Acad Sci. 2024;121:e2323052121
90. Basavalingappa RH, Massilamany C, Krishnan B, Gangaplara A, Kang G, Khalilzad-Sharghi V. et al. Identification of an Epitope from Adenine Nucleotide Translocator 1 That Induces Inflammation in Heart in A/J Mice. Am J Pathol. 2016;186:3160-75
91. Basavalingappa RH, Massilamany C, Krishnan B, Gangaplara A, Rajasekaran RA, Afzal MZ. et al. β1-Adrenergic Receptor Contains Multiple IAk and IEk Binding Epitopes That Induce T Cell Responses with Varying Degrees of Autoimmune Myocarditis in A/J Mice. Front Immunol. 2017;8:1567
92. Hapke N, Heinrichs M, Ashour D, Vogel E, Hofmann U, Frantz S. et al. Identification of a novel cardiac epitope triggering T-cell responses in patients with myocardial infarction. J Mol Cell Cardiol. 2022;173:25-9
93. Berner F, Bomze D, Lichtensteiger C, Walter V, Niederer R, Hasan Ali O. et al. Autoreactive napsin A-specific T cells are enriched in lung tumors and inflammatory lung lesions during immune checkpoint blockade. Sci Immunol. 2022;7:eabn9644
94. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y. et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med. 2016;375:1749-55
95. Watson RA, Ye W, Taylor CA, Jungkurth E, Cooper R, Tong O. et al. Severe acute myositis and myocarditis on initiation of 6-weekly pembrolizumab post-COVID-19 mRNA vaccination. J Immunother Cancer. 2024;12:e008151
96. Xia W, Zou C, Chen H, Xie C, Hou M. Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microRNA-34a/Kruppel-like factor 4 signaling. Cell Death Dis. 2020;11:575
97. Xia W, Chen H, Chen D, Ye Y, Xie C, Hou M. PD-1 inhibitor inducing exosomal miR-34a-5p expression mediates the cross talk between cardiomyocyte and macrophage in immune checkpoint inhibitor-related cardiac dysfunction. J Immunother Cancer. 2020;8:e001293
98. Wu M, Yang Y, Cai Y, Jiang S, Xiao H, Miao C. et al. Anti-CTLA-4 m2a Antibody Exacerbates Cardiac Injury in Experimental Autoimmune Myocarditis Mice By Promoting Ccl5-Neutrophil Infiltration. Adv Sci. 2024;11:e2400486
99. Zhou B, Qin Q, Fang Y, Liu X, Zhang M, Wang S. et al. Exosomes from human bone marrow MSCs alleviate PD-1/PD-L1 inhibitor-induced myocardial injury in melanoma mice by regulating macrophage polarization and pyroptosis. Life Sci. 2024;358:123108
100. Ju M, Zhang J, Deng Z, Wei M, Ma L, Chen T. et al. Prophylactic IL-23 blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. J Immunother Cancer. 2024;12:e009345
101. Chen Y, Luo Y, Liu Y, Luo D, Liu A. Dual efficacy of tocilizumab in managing PD-1 inhibitors-induced myocardial inflammatory injury and suppressing tumor growth with PD-1 inhibitors: a preclinical study. Cancer Immunol Immunother. 2025;74:52
102. Gergely TG, Kucsera D, Tóth VE, Kovács T, Sayour NV, Drobni ZD. et al. Characterization of immune checkpoint inhibitor-induced cardiotoxicity reveals interleukin-17A as a driver of cardiac dysfunction after anti-PD-1 treatment. Br J Pharmacol. 2023;180:740-61
103. Fu S, Guo Z, Xu X, Li Y, Choi S, Zhao P. et al. Protective effect of low-intensity pulsed ultrasound on immune checkpoint inhibitor-related myocarditis via fine-tuning CD4+ T-cell differentiation. Cancer Immunol Immunother. 2024;73:15
104. Bockstahler M, Fischer A, Goetzke CC, Neumaier HL, Sauter M, Kespohl M. et al. Heart-Specific Immune Responses in an Animal Model of Autoimmune-Related Myocarditis Mitigated by an Immunoproteasome Inhibitor and Genetic Ablation. Circulation. 2020;141:1885-902
105. Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. 2023;23:38-54
106. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736-46
107. Fa’ak F, Buni M, Falohun A, Lu H, Song J, Johnson DH. et al. Selective immune suppression using interleukin-6 receptor inhibitors for management of immune-related adverse events. J Immunother Cancer. 2023;11:e006814
108. Park E, Ciofani M. Th17 cell pathogenicity in autoimmune disease. Exp Mol Med. 2025;57:1913-27
109. Tang Z, Shi J, Xu S, Liu S, Yin W, Xiang Y. et al. Single-cell multiomics reveals macrophage-derived IL-23 and CXCL9/10 drive pathogenic IFNG+ IL17+ T cells in immunotherapy-related colitis. J Immunother Cancer. 2025;13:e011959
110. Salem J-E, Bretagne M, Abbar B, Leonard-Louis S, Ederhy S, Redheuil A. et al. Abatacept/Ruxolitinib and Screening for Concomitant Respiratory Muscle Failure to Mitigate Fatality of Immune-Checkpoint Inhibitor Myocarditis. Cancer Discov. 2023;13:1100-15
111. Jamilloux Y, El Jammal T, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev. 2019;18:102390
112. Dai C, Jiang M, Sun M-J. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2017;377:496-7
113. Bishu S, Melia J, Sharfman W, Lao CD, Fecher LA, Higgins PDR. Efficacy and Outcome of Tofacitinib in Immune checkpoint Inhibitor Colitis. Gastroenterology. 2021;160:932-934.e3
114. Sasson SC, Slevin SM, Cheung VTF, Nassiri I, Olsson-Brown A, Fryer E. et al. Interferon-Gamma-Producing CD8+ Tissue Resident Memory T Cells Are a Targetable Hallmark of Immune Checkpoint Inhibitor-Colitis. Gastroenterology. 2021;161:1229-1244.e9
115. Nguyen LS, Bretagne M, Arrondeau J, Zahr N, Ederhy S, Abbar B. et al. Reversal of immune-checkpoint inhibitor fulminant myocarditis using personalized-dose-adjusted abatacept and ruxolitinib: proof of concept. J Immunother Cancer. 2022;10:e004699
116. Xing Q, Zhang Z, Zhu B, Lin Q, Shen L, Li F. et al. Case Report: Treatment for steroid-refractory immune-related myocarditis with tofacitinib. Front Immunol. 2022;13:944013
117. Liu Q, Liu M, Zou Z, Lin J, Zhang N, Zhao L. et al. Tofacitinib for the treatment of immune-related adverse events in cancer immunotherapy: a multi-center observational study. J Transl Med. 2024;22:803
118. Mok S, Ağaç Çobanoğlu D, Liu H, Mancuso JJ, Allison JP. Post-immunotherapy CTLA-4 Ig treatment improves antitumor efficacy. Proc Natl Acad Sci. 2024;121:e2404661121
119. Salem J-E, Allenbach Y, Vozy A, Brechot N, Johnson DB, Moslehi JJ. et al. Abatacept for Severe Immune Checkpoint Inhibitor-Associated Myocarditis. N Engl J Med. 2019;380:2377-9
120. Liu S, Chan J, Brinc D, Gandhi S, Izenberg A, Delgado D. et al. Immune Checkpoint Inhibitor-Associated Myocarditis With Persistent Troponin Elevation Despite Abatacept and Prolonged Immunosuppression. JACC CardioOncology. 2020;2:800-4
121. Liu M, Wang X, Du X, Wu W, Zhang Y, Zhang P. et al. Soluble CTLA-4 mutants ameliorate immune-related adverse events but preserve efficacy of CTLA-4- and PD-1-targeted immunotherapy. Sci Transl Med. 2023;15:eabm5663
122. Baughman KL. Diagnosis of Myocarditis. Circulation. 2006;113:593-5
123. Ederhy S, Fenioux C, Cholet C, Rouvier P, Redheuil A, Cohen A. et al. Practical cardiovascular imaging approach to diagnose immune checkpoint inhibitor myocarditis. Eur Heart J - Cardiovasc Imaging. 2021;22:372-4
124. Jimenez J, Kostelecky N, Mitchell JD, Zhang KW, Lin C-Y, Lenihan DJ. et al. Clinicopathological classification of immune checkpoint inhibitor-associated myocarditis: possible refinement by measuring macrophage abundance. Cardio-Oncol. 2023;9:14
125. Champion SN, Stone JR. Immune checkpoint inhibitor associated myocarditis occurs in both high-grade and low-grade forms. Mod Pathol. 2020;33:99-108
126. Palaskas NL, Segura A, Lelenwa L, Siddiqui BA, Subudhi SK, Lopez-Mattei J. et al. Immune checkpoint inhibitor myocarditis: elucidating the spectrum of disease through endomyocardial biopsy. Eur J Heart Fail. 2021;23:1725-35
127. Chimenti C, Frustaci A, Chimenti C, Frustaci A. Contribution and Risks of Left Ventricular Endomyocardial Biopsy in Patients With Cardiomyopathies. Circulation. 2013;128:1531-41
128. Wheen P, Armstrong R, Daly CA. Recent Advances in T1 and T2 Mapping in the Assessment of Fulminant Myocarditis by Cardiac Magnetic Resonance. Curr Cardiol Rep. 2020;22:47
129. Ferreira VM, Schulz-Menger J, Holmvang G, Kramer CM, Carbone I, Sechtem U. et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation. J Am Coll Cardiol. 2018;72:3158-76
130. Thavendiranathan P, Zhang L, Zafar A, Drobni ZD, Mahmood SS, Cabral M. et al. Myocardial T1 and T2 Mapping by Magnetic Resonance in Patients With Immune Checkpoint Inhibitor-Associated Myocarditis. J Am Coll Cardiol. 2021;77:1503-16
131. Zhang L, Awadalla M, Mahmood SS, Nohria A, Hassan MZO, Thuny F. et al. Cardiovascular magnetic resonance in immune checkpoint inhibitor-associated myocarditis. Eur Heart J. 2020;41:1733-43
132. Quinaglia T, Gongora C, Awadalla M, Hassan MZO, Zafar A, Drobni ZD. et al. Global Circumferential and Radial Strain Among Patients With Immune Checkpoint Inhibitor Myocarditis. JACC Cardiovasc Imaging. 2022;15:1883-96
133. Tamura Y, Tamura Y, Takemura R, Yamada K, Taniguchi H, Iwasawa J. et al. Longitudinal Strain and Troponin I Elevation in Patients Undergoing Immune Checkpoint Inhibitor Therapy. JACC CardioOncology. 2022;4:673-85
134. Awadalla M, Mahmood SS, Groarke JD, Hassan MZO, Nohria A, Rokicki A. et al. Global Longitudinal Strain and Cardiac Events in Patients With Immune Checkpoint Inhibitor-Related Myocarditis. J Am Coll Cardiol. 2020;75:467-78
135. Caforio ALP, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB. et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34:2636-48
136. Lyon AR, López-Fernández T, Couch LS, Asteggiano R, Aznar MC, Bergler-Klein J. et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur Heart J. 2022;43:4229-361
137. Nensa F, Kloth J, Tezgah E, Poeppel TD, Heusch P, Goebel J. et al. Feasibility of FDG-PET in myocarditis: Comparison to CMR using integrated PET/MRI. J Nucl Cardiol. 2018;25:785-94
138. Tong J, Vogiatzakis N, Andres MS, Senechal I, Badr A, Ramalingam S. et al. Complementary use of cardiac magnetic resonance and 18 F-FDG positron emission tomography imaging in suspected immune checkpoint inhibitor myocarditis. Cardio-Oncol. 2024;10:53
139. Rischpler C, Rassaf T, Umutlu L, Herrmann K, Schlosser T-W, Totzeck M. Imaging the Inflammatory Response in Checkpoint Inhibition Myocarditis. J Nucl Med. 2022;63:14-6
140. Arponen O, Skyttä T. Immune checkpoint inhibitor-induced myocarditis not visible with cardiac magnetic resonance imaging but detected with PET-CT: a case report. Acta Oncol. 2020;59:490-2
141. Zhang X, Song W, Qin C, Lan X. Different displays of 13N-NH3 myocardial perfusion and cardiac 68Ga-FAPI PET in immune checkpoint inhibitor-associated myocarditis-induced heart failure. Eur J Nucl Med Mol Imaging. 2023;50:964-5
142. Boughdad S, Latifyan S, Fenwick C, Bouchaab H, Suffiotti M, Moslehi JJ. et al. 68Ga-DOTATOC PET/CT to detect immune checkpoint inhibitor-related myocarditis. J Immunother Cancer. 2021;9:e003594
143. Vasbinder A, Chen Y, Procureur A, Gradone A, Azam TU, Perry D. et al. Biomarker Trends, Incidence, and Outcomes of Immune Checkpoint Inhibitor-Induced Myocarditis. JACC CardioOncology. 2022;4:689-700
144. Qin Y, Zhang T, Du Z, Chen S, Li Y, Lv Y. et al. Prognosis of immune checkpoint inhibitor-related myocarditis: Retrospective experience of a single institution. Int Immunopharmacol. 2024;136:112385
145. Pavo N, Raderer M, Hülsmann M, Neuhold S, Adlbrecht C, Strunk G. et al. Cardiovascular biomarkers in patients with cancer and their association with all-cause mortality. Heart. 2015;101:1874-80
146. Petricciuolo S, Delle Donne MG, Aimo A, Chella A, De Caterina R. Pre-treatment high-sensitivity troponin T for the short-term prediction of cardiac outcomes in patients on immune checkpoint inhibitors. Eur J Clin Invest. 2021;51:e13400
147. Waissengein B, Abu Ata B, Merimsky O, Shamai S, Wolf I, Arnold JH. et al. The predictive value of high sensitivity troponin measurements in patients treated with immune checkpoint inhibitors. Clin Res Cardiol. 2023;112:409-18
148. Bonaca MP, Olenchock BA, Salem J-E, Wiviott SD, Ederhy S, Cohen A. et al. Myocarditis in the Setting of Cancer Therapeutics. Circulation. 2019;140:80-91
149. Rossi VA, Gawinecka J, Dimitriou F, von Eckardstein A, Dummer R, Ruschitzka F. et al. Value of troponin T versus I in the diagnosis of immune checkpoint inhibitor-related myocarditis and myositis: rechallenge? ESC Heart Fail. 2023;10:2680-5
150. Lehmann LH, Heckmann MB, Bailly G, Finke D, Procureur A, Power JR. et al. Cardiomuscular Biomarkers in the Diagnosis and Prognostication of Immune Checkpoint Inhibitor Myocarditis. Circulation. 2023;148:473-86
151. Drobni ZD, Zafar A, Zubiri L, Zlotoff DA, Alvi RM, Lee C. et al. Decreased Absolute Lymphocyte Count and Increased Neutrophil/Lymphocyte Ratio With Immune Checkpoint Inhibitor-Associated Myocarditis. J Am Heart Assoc. 2020;9:e018306
Author contact
![]() Corresponding authors: Dr. Libo Liu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing 100029, China, E-mail: liboliu2007com. Dr. Chenchen Tu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing 100029, China, E-mail: tcc2033ccmu.edu.cn. Dr. Xiantao Song, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing 100029, China, E-mail: song0929ccmu.edu.cn.
Corresponding authors: Dr. Libo Liu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing 100029, China, E-mail: liboliu2007com. Dr. Chenchen Tu, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing 100029, China, E-mail: tcc2033ccmu.edu.cn. Dr. Xiantao Song, Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing 100029, China, E-mail: song0929ccmu.edu.cn.