Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(8):2339-2349. doi:10.7150/thno.17555 This issue Cite
Research Paper
YD277 Suppresses Triple-Negative Breast Cancer Partially Through Activating the Endoplasmic Reticulum Stress Pathway
Zekun Chen1, 2, 3#, Qiuju Wu 4, 2#, Ye Ding5, Wenhui Zhou1, 2, Rong Liu2, Haiying Chen5, Jia Zhou5 ![]() , Jing Feng1
, Jing Feng1 ![]() , Ceshi Chen2
, Ceshi Chen2 ![]()
1. Department of Laboratory Medicine & Central Laboratory, Southern Medical University Affiliated Fengxian Hospital, Shanghai, China, 201499;
2. Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences and Yunnan Province, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China, 650223;
3. Department of Laboratory Medicine, Huizhou No.3 People's Hospital, Affiliated hospital of Guangzhou Medical University, Huizhou, Guangdong, China, 516002;
4. Fengxian District Center Hospital Graduate Student Training Base, Jinzhou Medical University, Shanghai, China, 201499;
5. Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas, 77555, United States.
# These authors contributed equally to this work.
Received 2016-9-13; Accepted 2017-4-16; Published 2017-6-11
Abstract

Triple-negative breast cancer (TNBC) is an aggressive malignancy with poor clinical outcomes. YD277 is a novel small molecule derived from ML264, a KLF5 inhibitor that elicits cytotoxic effects in colon cancer cell lines. Our previous studies suggest that Krüpple-like factor 5 (KLF5) is a promising therapeutic target for TNBC. In this study, we demonstrated that YD277 significantly induced G1 cell cycle arrest and apoptosis in MDA-MB-231 and MDA-MB-468 TNBC cells, independent of KLF5 inhibition. YD277 also reduced the protein expression levels of Cyclin D1, Bcl2 and Bclxl and promoted the expression of p21 and p27. Moreover, the pro-apoptotic activity of YD277 in TNBC was mediated by the transcription of IRE1α, a key molecule in the endoplasmic reticulum (ER) stress pathway. Finally, YD277 (15 mg/kg) significantly suppressed the growth of MDA-MB-231 tumor xenografts in nude mice. These findings indicate that YD277 is a promising chemotherapeutic candidate for TNBC.
Keywords: YD277, KLF5, Triple-Negative Breast Cancer, IRE1α, ER stress.
Introduction
Breast cancer is one of the most commonly diagnosed cancers and the second leading cause of cancer-related mortality in women. Indeed, breast cancer alone accounts for 29% of all new cancer cases in women [1]. Triple-negative breast cancer (TNBC) is a breast cancer subtype that lacks estrogen receptor (ERα), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) expression, and it accounts for 10-24% of invasive breast cancer cases. Compared to patients with other breast cancer subtypes, TNBC patients are usually younger and have poorer survival regardless of tumor stage [2]. Traditional targeted therapies have little success or even fail to benefit TNBC patients [3]. Given the lack of effective targeted therapeutics for TNBC patients, the development of new treatment regimens is urgently needed.
ML264 was originally identified as an inhibitor of Krüpple-like factor 5 (KLF5), an oncogenic transcription factor highly expressed in colorectal cancer [4]. It has been reported that ML264 potently inhibits KLF5 expression, thereby reducing the proliferation of colon cancer cell lines and the growth of xenografts in mice [4]. Our previous studies have demonstrated that KLF5 also promotes the proliferation, survival, migration and invasion of TNBC cells, indicating that KLF5 may be a therapeutic target in this breast cancer subtype [5-9]. Inhibition of KLF5 by small molecular inhibitors such as curcumin and mifepristone suppresses bladder and breast cancers [10-12]. Therefore, ML264 may inhibit the proliferation of TNBC.
Endoplasmic reticulum (ER) stress plays an important role in drug-induced apoptosis. The accumulation of misfolded glycoproteins in the ER, as well as stressors such as chemotherapeutics and hypoxia, trigger the unfolded protein response (UPR) to maintain ER homeostasis. PKR-like ER stress kinase (PERK), inositol-requiring transmembrane kinase and endonuclease 1α (IRE1α), and activating transcription factor 6 (ATF6) are three primary UPR sensors that lead to distinct downstream signaling pathways [13]. Upon activation, these proteins dissociate from immunoglobulin binding protein (BIP) to protect cells from apoptosis [14]. However, prolonged ER stress induces apoptosis to eliminate damaged cells [15, 16]. Apoptosis is mediated by transcriptional activation of C/EBP homologous protein (CHOP) and c-JUN NH2-terminal kinase (JNK). In addition, cleaved caspase-3 is the executioner caspase in ER stress-mediated apoptosis [17].
In this study, we synthesized 24 novel derivatives of ML264. YD277 was identified as the most potent derivative, demonstrating at least 10-fold enhanced efficacy in TNBC cell lines compared to ML264. Interestingly, YD277 did not appear to function through inhibition of KLF5 expression in breast cancer cells. In addition, YD277 remarkably decreased the protein levels of Bcl2, Bclxl and CyclinD1 and activated the ER stress pathway. Furthermore, knockdown of IRE1α significantly reduced YD277-induced apoptosis. These findings suggest that YD277 has potential as a therapeutic agent for TNBC.
Materials and Methods
Cell lines and reagents
All cell lines used in this study were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia, USA) and validated by Short Tandem Repeat (STR) analysis. MCF7 and HCT116 cells were maintained in minimum essential medium (MEM), and McCoy's medium, respectively. MDA-MB-231 cells were cultured Ham's F12. SUM149PT cells were cultured Ham's F12 with 5 μg/ml insulin and 500 ng/ml hydrocortisone. MDA-MB-468 and NIH3T3 cells were cultured in DMEM (high glucose), whereas T-47D, MDA-MB-453, Hs578T, HCC1806, HC11 and HCC1937 cells were maintained in RPMI-1640. MCF10A cells were cultured in DMEM/F12 with 5 μg/ml insulin and 10 ng/ml cholera toxin. All media were supplemented with 1% penicillin/streptomycin and 10% FBS.
YD277 and other novel derivatives were designed and synthesized in the laboratory of Dr. Jia Zhou at the University of Texas Medical Branch through the chemical modification of various ML264 pharmacophoric moieties. The reference compound, ML264 [18], was also synthesized by Dr. Zhou's group. The structures of these compounds are listed in Supplementary Table 1. YD277 was dissolved in DMSO and diluted in normal saline for cell culture studies.
Cell viability and proliferation assays
Sulforhodamine B (SRB, Sigma, St. Louis, MO) assays were performed to measure cell viability. HCT116, MDA-MB-231 and HCC1806 cells were seeded in 96-well plates at 4000 cells/well. After 24 h, the cells were treated with ML264 or its 24 novel derivatives at a concentration of 10 μM. DMSO was used as the negative control. To measure the half maximal inhibitory concentration (IC50), cancer cells were seeded in 48-well plates at 2.5×104 cells/well. Twenty-four hours later, the cells were treated with different concentrations of YD277 (10-0.3 μM) for 48 h. The cells were then fixed with 100-200 μl of 10% trichloroacetic acid (TCA, Sigma) for 60 min, washed 5 times with deionized water and air dried. The cells were then stained with 50-100 μl of 0.4% (w/v) SRB in acetic acid for 5 min. Then, the plates were washed 5 times with 1% acetic acid and dried. Finally, 100-200 μl of 10 mM Tris base was added to each well. Optical densities at 530 nm were measured using an Epoch microplate reader (BioTek, Winooski, VT, USA). Each experiment was performed in triplicate and repeated.
DNA synthesis was detected in MDA-MB-231 and MDA-MB-468 cells with the Click-iT EdU Microplate Assay kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Ten fields for each sample were randomly captured. EdU-positive and DAPI-positive cells were counted using Image-Pro Plus 6.0 software (IPP, Media Cybernetics, Inc.).
Cell cycle and apoptosis analyses
MDA-MB-231 and MDA-MB-468 cells were seeded in 12-well plate at 2.5×105 cells/well. After 24 h, the cells were treated with YD277 at 1, 3 and 10 μM for 36 h. For cell cycle analyses, harvested cells were suspended in 200 μl PBS with 0.6% NP-40, 2 mg/ml RNase, and 0.1 mg/ml propidium iodide (PI, eBioscience, San Diego, CA, USA) for 30 min at 37°C in dark. Alternatively, the cells were stained with Annexin V and PI for apoptosis analyses. All cells were analyzed on an Accuri C6 flow cytometer (BD, San Jose, CA, USA).
Western blot (WB)
MDA-MB-231 and MDA-MB-468 cells were treated with YD277 in 6-well plates. The cells then were collected in lysis buffer containing protease inhibitors (P8340, Sigma). The cell lysates were centrifuged and the supernatants were collected, mixed with sample loading buffer and boiled for 5 min. The samples were then subjected to SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were incubated with diluted primary antibodies (Supplementary Table 2) and HRP-conjugated secondary antibodies (eBioscience). Western Lighting Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Shelton, CT, USA) and an ImageQuant LAS 4000 biomolecular imager (GE Healthcare) were used to visualize the expression levels of specific proteins [19].
RNA interference
Cells were transfected with 50 nm of siRNA oligos (RiboBio Co. Ltd., Guangzhou, China) using LipofectamineTM 2000 (Invitrogen) according to the manufacturer's instructions. A Luc siRNA was used as the negative control. Twenty-four hours after transfection, the cells were treated with different concentrations of YD277 (0.3-10 μM) for 48 h.
Tumorigenesis
Animal studies were approved by the institutional ethics committee of Kunming Institute of Zoology, Chinese Academy of Sciences. Eighteen nude mice were purchased from SJA Lab Animal Co., Ltd. (Changsha, China) and were housed in an SPF animal facility. MDA-MB-231 cancer cells were injected subcutaneously into the left and right flanks of 6-week old female nude mice (1×106 cells/spot). Tumor volumes were measured with calipers and calculated using established methods [20]. When tumors reached a volume of approximately 10-20 mm3, the mice were randomly divided into two groups. Ten mice were treated with YD277 (15 mg/kg) and 8 mice were treated with the vehicle solution (sesame oil) as the negative control. All mice were injected intraperitoneally (i.p.) every two days. In addition, the mice were weighed and the tumor volumes were measured every 4 days. This experiment was terminated when tumor maximum size reached approximately 1 cm in diameter, at which point the tumors were isolated and weighed.
Statistical analysis
Statistical analyses were conducted with GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA). All data are presented as the mean ± SD. Differences between groups were identified using Student's t-test. p<0.05 was considered statistically significant.
Results
YD277 is the most potent anti-cancer compound among the tested ML264 derivatives
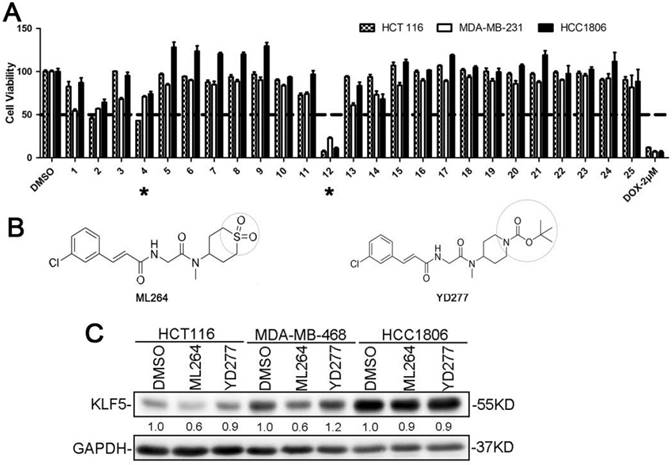
To develop novel potential drugs for KLF5-positive basal-like breast cancer, we synthesized a series of ML264 derivatives by chemically modifying its various pharmacophoric moieties. We then compared the cytotoxicity of these derivatives to that of ML264 in the colon cancer cell line, HCT116 (KLF5-positive), and two breast cancer cell lines, including MDA-MB-231 (KLF5-negative) and HCC1806 (KLF5-positive). All cells were treated with each compound (10 μM) for 48 h, and an SRB assay was then performed. Of the 24 derivatives, YD277 (#12) exhibited the most potent cytotoxicity in all three cancer cell lines regardless of KLF5 status (Fig. 1A). YD277 was also more potent than the reference compound, ML264 (#4, Fig. 1B), and had similar effects to the chemotherapeutic drug, doxorubicin, in these cancer cells (Fig. 1A).
YD277 is the most potent anti-cancer compound among the tested ML264 derivatives A. HCT116, MDA-MB-231 and HCC1806 cells were treated with each compound (10 μM) for 48 h and an SRB assay was performed. DMSO and Doxorubicin were used as the negative and positive controls, respectively, and ML264 (#4) was used as the reference compound. YD277 (#12) exhibited the greatest cytotoxicity among all of the compounds tested. B. The molecular structures of ML264 and YD277. C. YD277 did not inhibit KLF5 expression in cancer cells. Three KLF5-positive cancer cell lines (HCT116, MDA-MB-468 and HCC1806) were treated with ML264 (10 μM) or YD277 (5 μM) for 24 h. The expression levels of KLF5 protein were measured by WB and GAPDH normalized quantitative data were shown below the blot. ML264 inhibited KLF5 protein expression in HCT116 and MDA-MB-468 cells.
We next measured IC50 of YD277 in 13 different cell lines, including 7 TNBC cell lines, 2 ERα-positive breast cancer cell lines, 1 colon cancer cell line, and 3 control cell lines. The IC50 values in all cancer cell lines are located within the 1.5-10 μM range, indicating that YD277 does not exhibit cell type specificity. Immortalized MCF10A breast epithelial cells were also sensitive to YD277 (IC50 = 8.5 μM), whereas normal HC11 breast epithelial cells [21] and NIH-3T3 mouse fibroblasts were more resistant (IC50 = 20 μM and 35 μM, respectively) than the cancer cell lines (Table 1). The IC50 values of ML264 in most of these cell lines were greater than those of YD277 (Supplementary Table 3).
IC50 of YD277 in 13 different cell lines, including 7 TNBC cell lines, 2 ERα-positive breast cancer cell lines, 1 colorectal cancer cell line, and 3 control cell lines.
| Types | Cell lines | IC50 (μM) |
|---|---|---|
| ERα+ breast cancer | MCF-7 | 1.5 |
| T-47D | 2 | |
| TNBC | SUM149PT | 2.5 |
| MDA-MB-231 | 2 | |
| MDA-MB-453 | 6 | |
| MDA-MB-468 | 2 | |
| Hs578T | 6 | |
| HCC1806 | 9 | |
| HCC1937 | 2 | |
| Normal | MCF10A | 8.5 |
| HC11 | 20 | |
| NIH3T3 | 35 | |
| Colorectal cancer | HCT116 | 10 |
YD277 decreases cancer cell viability in a KLF5-independent manner
Our previous studies suggest that KLF5 is a target in basal-like breast cancer cell lines [5-9]. ML264 was originally developed as a KLF5 inhibitor [18]. However, the ML264 derivative YD277 exhibited more potent cytotoxicity in both KLF5-positive (SUM149PT, MDA-MB-453, MDA-MB-468, HCC1937, HCC1806, HCT116 and MCF10A) and KLF5-negative (MCF7, T-47D, MDA-MB-231 and Hs578T) cell lines, indicating that YD277 functions independently of KLF5 expression status.
To test whether YD277 inhibits KLF5 in a manner similar to that of ML264, we treated three KLF5-positive cancer cell lines, HCT116, MDA-MB-468 and HCC1806, with ML264 and YD277 for 24 h and measured KLF5 protein expression levels by WB. As shown in Fig. 1C, ML264 reduced the protein expression levels of KLF5 in HCT116 cells, which is consistent with a previous study [4]. In breast cancer cell lines, ML264 reduced the expression levels of KLF5 protein in MDA-MB-468 cells, but not in HCC1806 cells. These results suggest that ML264 reduces KLF5 expression in a cell line-specific manner. Importantly, YD277 did not decrease KLF5 protein levels in HCT116, MDA-MB-468 or HCC1806 cells (Fig. 1C), suggesting that YD277 decreases cancer cell viability independently of reducing KLF5 expression.
YD277 inhibits DNA synthesis in MDA-MB-231 and MDA-MB-468 TNBC cells
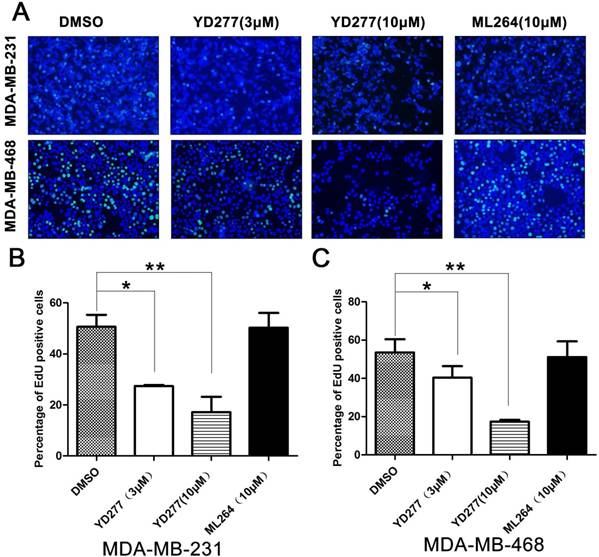
To determine whether YD277 inhibits cancer cell proliferation, we performed an EdU assay to visualize the effects of YD277 on DNA synthesis in MDA-MB-231 and MDA-MB-468 cells. We selected these two TNBC cancer cell lines because they are relatively more sensitive to YD277. MDA-MB-231 represents the KLF5-negative claudin-low triple-negative subtype, whereas MDA-MB-468 represents the KLF5-positive basal-like subtype. YD277 (3 μM and 10 μM) significantly decreased DNA synthesis in both cancer cell lines in a dose-dependent manner (Fig. 2). However, the reference compound ML264 (10 μM) did not decrease DNA synthesis in either cancer cell lines (Fig. 2).
YD277 inhibits DNA synthesis in MDA-MB-231 and MDA-MB-468 TNBC cells A. MDA-MB-231 and MDA-MB-468 cells were treated with YD277 (3 and 10 μM) for 24 h. DMSO and ML264 (10 μM) were used as controls. DNA synthesis was measured using an EdU incorporation assay. B. Quantitative data for the EdU incorporation assay in MDA-MB-231 cells. Percentages of EdU-positive proliferating cells out of total cells are presented. Data represent the mean ± SD (n=3). *p < 0.05, **p < 0.01. C. Quantitative data for the EdU incorporation assay in MDA-MB-468 cells.
YD277 induces G1 cell cycle arrest in MDA-MB-231 and MDA-MB-468 cells
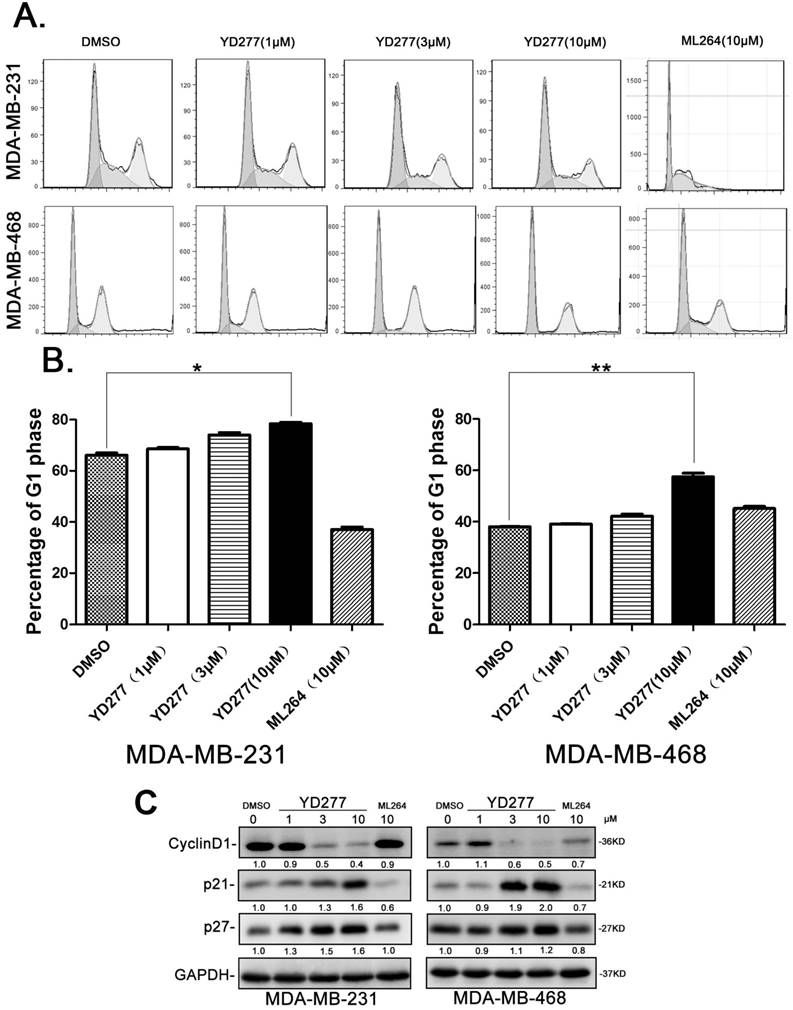
Given that YD277 inhibits cancer cell DNA synthesis, we wondered whether YD277 causes cell cycle arrest. We treated MDA-MB-231 and MDA-MB-468 cells with YD277 (1, 3, and 10 μM) for 36 h and analyzed their cell cycle profiles by PI staining. As shown in Fig. 3A-B, in both cancer cell lines, YD277 (10 μM) significantly increased the percentage of cells in G1 phase compared to cells treated with DMSO. In agreement with the cell cycle results, YD277 dramatically reduced the expression levels of Cyclin D1 and increased the expression levels of p21 and p27 (Fig. 3C). By contrast, ML264 (10 μM) did not induce G1 cell cycle arrest and change the expression of these cell cycle regulatory proteins (Fig. 3).
YD277 induces G1 cell cycle arrest in MDA-MB-231 and MDA-MB-468 cells A. The cell cycle distributions of MDA-MB-231 and MDA-MB-468 cells after YD277 treatment. The cells were treated with YD277 at 1, 3 and 10 μM as indicated for 36 h. The cells were stained with propidium iodide (PI) for 30 min at 37°C in dark and subjected to flow cytometer analysis. ML264 was used as the control, which induced S and G2/M phase arrest. B. Compared to DMSO and ML264, YD277 (10 μM) significantly increased the percentage of MDA-MB-231 and MDA-MB-468 cells in G1 phase. Each experiment was conducted in triplicate and data are presented as the mean ± SD (n=3). *p < 0.05, **p < 0.01. C. MDA-MB-231 and MDA-MB-468 cells were treated with YD277, ML264, or DMSO for 36 h, and the expression levels of cell cycle related proteins, such as Cyclin D1, p21, and p27, were detected by WB. GAPDH normalized quantitative data were shown below each blot.
YD277 induces apoptosis in MDA-MB-231 and MDA-MB-468 cells
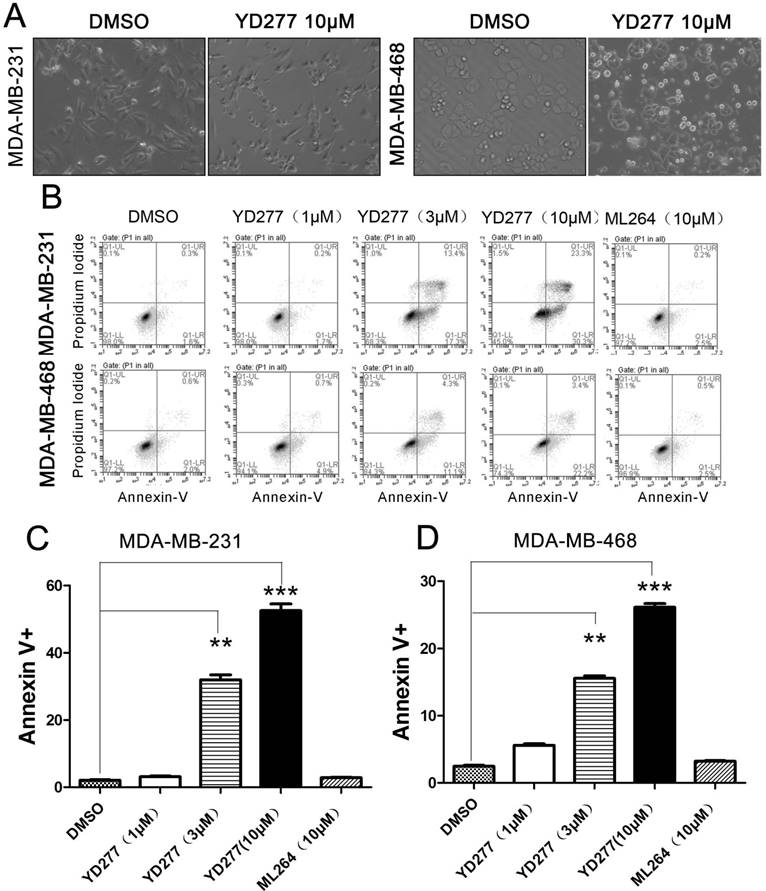
YD277 (10 μM) induced considerable apoptosis (Fig. 4A). To test whether YD277 induces apoptosis in TNBC cell lines, we treated MDA-MB-231 and MDA-MB-468 cells with YD277 (1-10 μM) for 36 h and stained the cells with Annexin V and PI. By flow cytometry, YD277 significantly increased the proportion of Annexin V-positive apoptotic cells in a dose-dependent manner (Fig. 4B-D). These data indicate that YD277 induces apoptosis in TNBC cells.
YD277 induces apoptosis in MDA-MB-231 and MDA-MB-468 TNBC cells A. The morphology of MDA-MB-231 and MDA-MB-468 cells changed dramatically after 36 h of treatment with YD277 (10 μM). B. YD277 induces apoptosis in MDA-MB-231 and MDA-MB-468 cells in a dose-dependent manner as measured by Annexin V staining and flow cytometry analysis. C. Proportions of Annexin V-positive MDA-MB-231 cells. The data are presented as the mean ± SD (n=3). **p < 0.01, ***p < 0.001. D. Proportions of Annexin V-positive MDA-MB-468 cells.
YD277 regulates the expression levels of apoptosis-related proteins
To investigate the mechanism by which YD277 induces apoptosis, we examined the expression levels of apoptosis-related proteins by WB. As shown in Fig. 5A, in both MDA-MB-231 and MDA-MB-468 cells, YD277 dramatically reduced the expression levels of Bcl-2 and Bcl-xl and increased the expression levels of cleaved Caspase-9, 7, 3 and cleaved PARP in a dose-dependent manner. When we treated MDA-MB-231 cells with the caspase inhibitor zVAD, YD277-induced cell viability loss was significantly but not completely blocked (Fig. S1), suggesting that caspase activation plays an important role in YD277 induced apoptosis.
YD277 regulates the expression levels of apoptosis-related proteins and activates the ER stress pathway A. MDA-MB-231 and MDA-MB-468 cells were treated with YD277, ML264, or DMSO for 36 h, and the expression levels of apoptosis-related proteins were detected by WB. GAPDH normalized quantitative data for Bcl2 and Bcl-xL were shown below their panel. B. MDA-MB-231 and MDA-MB-468 cells were treated with drugs for 36 h, and WB was performed to detect the expression levels of proteins in the ER stress pathway. GAPDH normalized quantitative data were shown below their panel. P-JNK and p-c-Jun were normalized to JNK and c-Jun, respectively. C. IRE1α depletion dramatically increased the IC50 values of YD277 in both MDA-MB-231 and MDA-MB-468 cells. The cells were transiently transfected with 50 nM siRNA against IRE1α for 24 h and subsequently treated with YD277 for 48 h. Cell viability was measured using the SRB assay. D. Knockdown of IRE1α blocked YD277 (4 μM)-induced apoptosis as measured by Annexin V staining and flow cytometry analysis. Data are presented as the mean ± SD (n=3). **p < 0.01. E. IRE1α depletion in both MDA-MB-231 and MDA-MB-468 cells abolished YD277-induced PARP cleavage and JNK activation, as measured by WB. GAPDH normalized quantitative data were shown below their panel. p-JNK was normalized to JNK.
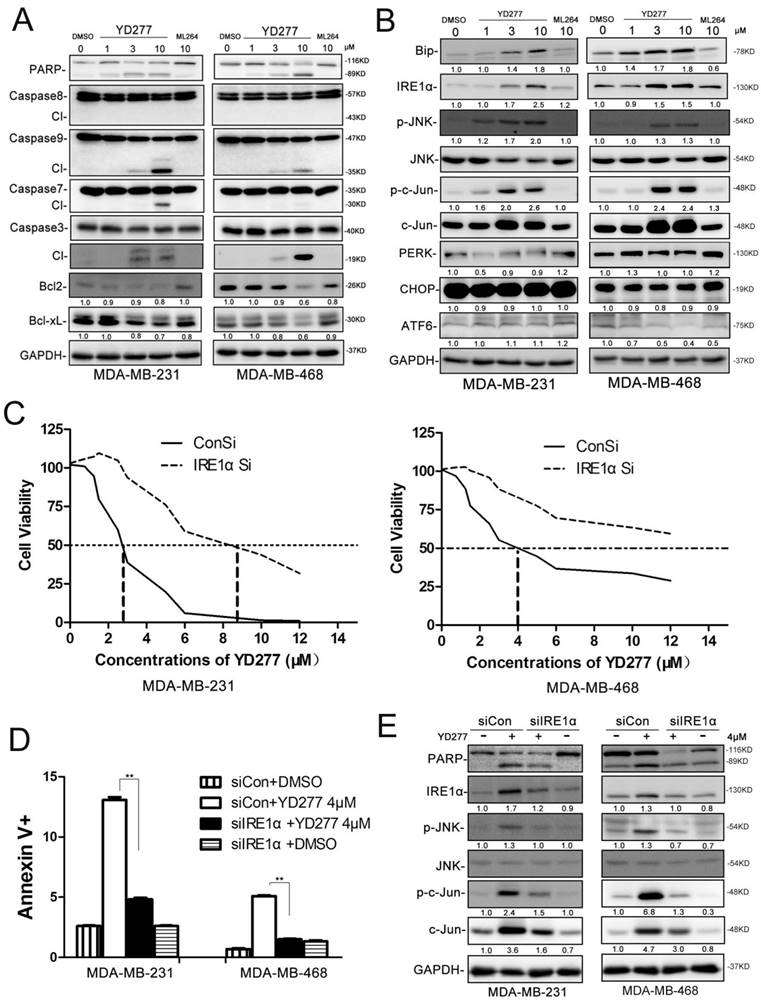
We noticed that YD277 did not cause the cleavage of caspase-8 (Fig. 5A), an initial caspase in the extrinsic apoptotic pathway. We did not observe any cleavage of BID (Fig. S2). DR4/5 and Bak/Bax were not up-regulated by YD277 (Fig. S2). To further test whether YD277 induces apoptosis through the intrinsic apoptotic pathway, we overexpressed Bcl2/Bclxl in MDA-MB-231 cells and treated the cells with YD277. As shown in Fig. S3, YD277-induced cell viability loss was significantly but not completely blocked, suggesting that loss of Bcl2/Bclxl plays an important role in YD277 induced apoptosis. This is consistent with the activation of caspase-9, an initial caspase in the intrinsic apoptotic pathway.
YD277 induces apoptosis through activating IRE1α, a key mediator in the ER stress pathway
It is well known that activation of the ER stress pathway by small molecules may induce cancer cell apoptosis [22]. Therefore, we treated MDA-MB-231 and MDA-MB-468 cells with YD277 and examined a number of proteins related to ER stress (Fig. 5B). Interestingly, BIP, IRE1α, pJNK, p-c-Jun, and c-Jun were dramatically induced by YD277 in a dose-dependent manner, whereas PERK, CHOP and ATF6 exhibited no significant changes (Fig. 5B).
To test whether YD277 causes apoptosis through IRE1α, we knocked down IRE1α in MDA-MB-231 and MDA-MB-468 cells. In the absence of IRE1α, the IC50 value was increased to greater than 8.5 μM (Fig. 5C). In the absence of IRE1α, YD277-induced apoptosis was almost completely blocked (Fig. 5 D-E), suggesting that IRE1α plays a crucial role in YD277-induced apoptosis.
To characterize the mechanism by which YD277 induces the IRE1α expression, we treated MD-MB-231 cells with YD277 and measured the IRE1α mRNA levels by RT-qPCR. As shown in Fig. S5A-B, YD277 significantly increased the IRE1α mRNA levels in time and dose dependent manners. Furthermore, we demonstrated that actinomycin D completely blocked the induction of IRE1α by YD277 (Fig. S5C), suggesting that YD277 up-regulates IRE1α at the transcriptional level.
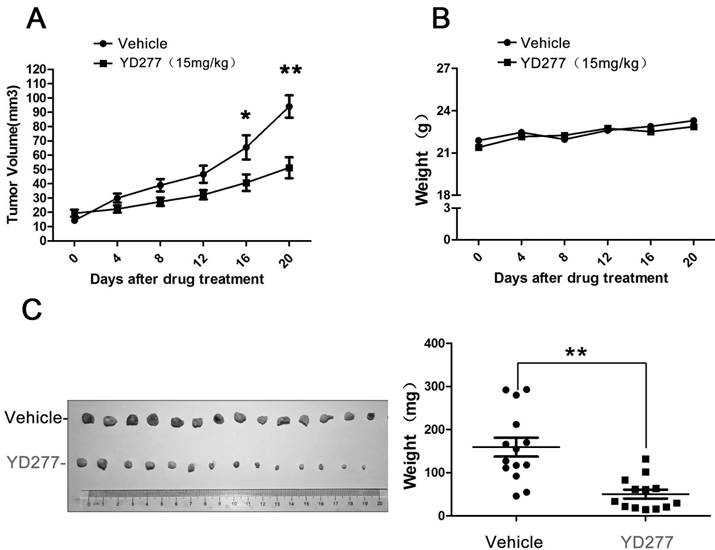
YD277 significantly inhibits the growth of MDA-MB-231 tumor xenografts in nude mice
Since YD277 suppressed TNBC cell proliferation and survival in vitro, we investigated its ability to suppress the growth of MDA-MB-231 tumor xenografts in nude mice. As shown in Fig. 6, YD277 (15 mg/kg) significantly suppressed the growth of MDA-MB-231 xenografts over the course of 20 days (Fig. 6A) compared to the vehicle control. In addition, YD277 did not affect mouse body weight (Fig. 6B). Tumors were harvested at the end of treatment (Fig. 6C), and an analysis of tumor weights indicated that YD277 significantly decreased tumor weights compared to the vehicle control.
YD277 significantly inhibits the growth of MDA-MB-231 tumor xenografts in nude mice A. Over the course of 20 days, YD277 (15 mg/kg) significantly suppressed the growth of MDA-MB-231 tumor xenographs compared to the vehicle control. B. YD277 did not reduce mouse body weights. C. Photographs of the tumors collected following 20 days of treatment with vehicle or YD277. Tumor weights are also presented (n=16). **p < 0.01. Only 14 tumors are shown after we removed outliers.
Discussion
TNBC is an aggressive malignancy with a poor prognosis than other breast cancer subtypes. Given that TNBC does not respond to endocrine therapy or other available targeted treatments, the development of more effective and safer anti-cancer drugs for TNBC is urgently needed. KLF5 is a therapeutic target in TNBC and bladder cancer because KLF5 promotes cancer cell proliferation, survival, migration and invasion. ML264 was identified as a KLF5 inhibitor that suppresses tumor growth of colorectal cancer. However, we found that the ability of ML264 to suppress TNBC was limited. It is imperative to develop ML264 derivatives, particularly for TNBC. Thus, we synthesized 24 new ML264 derivatives to identify potential anti-TNBC compounds.
In the present study, YD277 was identified as the most effective inhibitor against breast cancer among the tested ML264 derivatives. Unlike ML264, which leads to a significant increase in S-phase arrest in DLD-1 and HCT116 cells [18], YD277 suppressed TNBC cells by inducing G1-phase cell cycle arrest and reducing DNA synthesis. Consistent with these results, we observed that YD277 decreased Cyclin D1 expression and increased the expression of p21 and p27. It is well known that Cyclin D1 plays a key role in promoting the G1-S cell cycle transition. Additionally, YD277 reduced the expression of the anti-apoptotic proteins Bcl2 and Bclxl and induced apoptosis in TNBC cells. To our surprise, YD277, as a derivative of ML264, did not affect KLF5 but nevertheless exhibited an enhanced capacity to suppress TNBC cell lines. We also demonstrated that YD277 activates the ER stress pathway, namely by upregulating the expression of IRE1α, which is responsible for apoptosis. Finally, YD277 significantly inhibits tumor growth in vivo without reducing mouse body weight.
ER is the organelle in which secretory proteins are synthesized and folded. Perturbation of ER homeostasis affects protein folding and causes ER stress, shifting the cell to a pro-survival state. However, prolonged ER stress induces apoptosis to eliminate damaged cells. IRE1α has dual enzymatic activities, consisting of a serine/threonine kinase domain plus a C-terminal ribonuclease (RNase) domain. Upon ER stress, IRE1α RNase domain is activated to degrades ER-bound mRNAs, a process referred to as regulated IRE1-dependent decay that leads to cell death [23]. It has been previously shown that activated IRE1α on the ER membrane recruits TNF receptor-associated factor 2 (TRAF2), thus activating JNK and inducing cell death [10, 24]. In addition, overexpression of IRE1α induces apoptosis in HeLa cells [25]. YD277 activates ER stress-mediated apoptosis in TNBC cell lines by significantly inducing IRE1α transcription, and it led to JNK activation in a dose-dependent manner. We also show that TNBC cell lines were resistant to YD277-induced ER stress and apoptosis when IRE1α was depleted by siRNA. Thus, IRE1α plays a key role in YD277-induced apoptosis. Additionally, we found that YD277 also inhibited the activation of AKT (Fig. S4). Nevertheless, the direct target of YD277 and the mechanism by which YD277 induces IRE1α expression is still unknown and requires further investigation.
Our study shows that YD277 has no effect on mouse body weight after 20 days of treatment. This result may suggest that YD277 has low toxicity. However, detailed investigations of in vivo YD277 toxicity should be undertaken in the future.
In summary, we found that the novel ML264 derivative YD277 inhibits TNBC cell proliferation and tumor growth in nude mice and induces G1 cell cycle arrest and apoptosis. This compound exhibits a more than 10-fold increase in efficacy compared to the reference compound ML264. The mechanism by which YD277 inhibits TNBC may involve the induction of IRE1α, p-JNK, p-c-Jun, p21, and p27, activation of caspase-9, 7, 3, as well as the down-regulation of Cyclin D1, Bcl2, and Bclxl. The induction of IRE1α is essential for YD277-mediated apoptosis in TNBC. Therefore, YD277 has potential as a candidate drug for human TNBC and other cancers.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Prof. Qinhua Cui from Yunnan University for providing the Bcl2 and Bclxl expression vectors. This work was supported by grants from "Personalized Medicines - Molecular Signature-based Drug Discovery and Development," a Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12010303 to Chen, C.); The Shanghai Health System Outstanding Academic Leader Training Program (XBR2013114 to Feng, J.); and the National Natural Science Foundation of China (U1602221 and 81325016 to Chen, C., and 81672624 to Feng, J.). In addition, Zhou, J. received support from the National Institutes of Health (P30 DA028821 and R01 DA038446) and a Cancer Prevention Research Institute of Texas (CPRIT) award.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5-29
2. Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109:1721-8
3. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016
4. Ruiz de Sabando A, Wang C, He Y, Garcia-Barros M, Kim J, Shroyer KR. et al. ML264, A Novel Small-Molecule Compound That Potently Inhibits Growth of Colorectal Cancer. Mol Cancer Ther. 2016;15:72-83
5. Jia L, Zhou Z, Liang H, Wu J, Shi P, Li F. et al. KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene. 2016;35:2040-51
6. Chen C, Benjamin MS, Sun X, Otto KB, Guo P, Dong XY. et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. International journal of cancer. 2006;118:1346-55
7. Liu R, Zheng HQ, Zhou Z, Dong JT, Chen C. KLF5 promotes breast cell survival partially through fibroblast growth factor-binding protein 1-pERK-mediated dual specificity MKP-1 protein phosphorylation and stabilization. The Journal of biological chemistry. 2009;284:16791-8
8. Zheng HQ, Zhou Z, Huang J, Chaudhury L, Dong JT, Chen C. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene. 2009;28:3702-13
9. Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cellular and molecular life sciences: CMLS. 2009;66:2691-706
10. Gao Y, Ding Y, Chen H, Chen H, Zhou J. Targeting Kruppel-like factor 5 (KLF5) for cancer therapy. Curr Top Med Chem. 2015;15:699-713
11. Liu R, Shi P, Nie Z, Liang H, Zhou Z, Chen W. et al. Mifepristone Suppresses Basal Triple-Negative Breast Cancer Stem Cells by Down-regulating KLF5 Expression. Theranostics. 2016;6:533-44
12. Gao Y, Shi Q, Xu S, Du C, Liang L, Wu K. et al. Curcumin promotes KLF5 proteasome degradation through downregulating YAP/TAZ in bladder cancer cells. International journal of molecular sciences. 2014;15:15173-87
13. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular cell biology. 2007;8:519-29
14. Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell death and differentiation. 2006;13:363-73
15. Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis: an international journal on programmed cell death. 2002;7:335-45
16. Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nature cell biology. 2001;3:E255-63
17. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell death and differentiation. 2004;11:381-9
18. Ruiz de Sabando A, Wang C, He Y, Garcia-Barros M, Kim J, Shroyer KR. et al. ML264 - a novel small-molecule compound that potently inhibits growth of colorectal cancer. Molecular cancer therapeutics. 2015;15(1):72-83
19. Kong Y, Chen J, Zhou Z, Xia H, Qiu MH, Chen C. Cucurbitacin E induces cell cycle G2/M phase arrest and apoptosis in triple negative breast cancer. PloS one. 2014;9:e103760
20. Qin J, Zhou Z, Chen W, Wang C, Zhang H, Ge G. et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nature communications. 2015;6:8471
21. Zhao D, Ma G, Zhang XL, He Y, Li M, Han XY. et al. Zinc Finger Homeodomain Factor Zfhx3 Is Essential for Mammary Lactogenic Differentiation by Maintaining Prolactin Signaling Activity. J Biol Chem. 2016;291:12809-20
22. Liu X, Guo S, Liu X, Su L. Chaetocin induces endoplasmic reticulum stress response and leads to death receptor 5-dependent apoptosis in human non-small cell lung cancer cells. Apoptosis. 2015;20:1499-507
23. Jiang D, Niwa M, Koong AC. Targeting the IRE1alpha-XBP1 branch of the unfolded protein response in human diseases. Semin Cancer Biol. 2015;33:48-56
24. Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K. et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Gene Dev. 2002;16:1345-55
25. Iwawaki T, Hosoda A, Okuda T, Kamigori Y, Nomura-Furuwatari C, Kimata Y. et al. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nature cell biology. 2001;3:158-64
Author contact
![]() Corresponding authors: Ceshi Chen, Tel: 86-871-5181944, E-mail: chenckiz.ac.cn; Jing Feng, E-mail: fengjing8801530com; or Jia Zhou, Tel: 1-409-7729748, E-mail: jizhouedu
Corresponding authors: Ceshi Chen, Tel: 86-871-5181944, E-mail: chenckiz.ac.cn; Jing Feng, E-mail: fengjing8801530com; or Jia Zhou, Tel: 1-409-7729748, E-mail: jizhouedu