Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Background
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(9):2452-2462. doi:10.7150/thno.18813 This issue Cite
Research Paper
miR-196a Enhances Neuronal Morphology through Suppressing RANBP10 to Provide Neuroprotection in Huntington's Disease
Lu-Shiun Her1*, Su-Han Mao2*, Chih-Yi Chang2*, Pei-Hsun Cheng2, Yu-Fan Chang2, Han-In Yang2, Chuan-Mu Chen4, Shang-Hsun Yang2, 3 ![]()
1. Department of Life Sciences, College of Bioscience and Biotechnology;
2. Department of Physiology, College of Medicine;
3. Institute of Basic Medical Sciences, National Cheng Kung University, Tainan 70101, Taiwan;
4. Department of Life Sciences, Agricultural Biotechnology Center, National Chung Hsing University, Taichung 40227, Taiwan.
* These authors contributed equally to this paper
Received 2016-12-18; Accepted 2017-4-18; Published 2017-6-24
Abstract

MicroRNAs (miRNAs) play important roles in several neurobiological processes, including the development and progression of diseases. Previously, we identified that one specific miRNA, miR-196a, provides neuroprotective effects on Huntington's disease (HD), although the detailed mechanism is still unclear. Based on our bioinformatic analyses, we hypothesize miR-196a might offer neuroprotective functions through improving cytoskeletons of brain cells. Here, we show that miR-196a could enhance neuronal morphology, further ameliorating intracellular transport, synaptic plasticity, neuronal activity, and learning and memory abilities. Additionally, we found that miR-196a could suppress the expression of RAN binding protein 10 (RANBP10) through binding to its 3' untranslated region, and higher expression of RANBP10 exacerbates neuronal morphology and intracellular transport. Furthermore, miR-196a enhances neuronal morphology through suppressing RANBP10 and increasing the ability of β-tubulin polymerization. Most importantly, we observed higher expression of RANBP10 in the brains of HD transgenic mice, and higher expression of RANBP10 might exacerbate the pathological aggregates in HD. Taken together, we provide evidence that enhancement of neuronal morphology through RANBP10 is one of the neuroprotective mechanisms for miR-196a. Since miR-196a has also been reported in other neuronal diseases, this study might offer insights with regard to the therapeutic use of miR-196a in other neuronal diseases.
Keywords: miR-196a, Neuronal morphology, RANBP10, Huntington's disease, β-tubulin polymerization, Neuroprotection.
Background
Huntington's disease (HD) is a human inheritable autosomal dominant disease, and this is caused by a mutation of CAG trinucleotide repeats in the exon 1 region of Huntingtin (HTT) gene [1]. The clinical symptoms of HD include cognitive dysfunction, emotional changes, mental deterioration, motor deficits, chorea and dystonia [2, 3]. To date, there is no cure, and thus HD patients suffer unimaginable pain until death. Mutant HTT is involved in abnormal gene regulation, and post-transcriptional regulation of non-coding RNAs, especially microRNAs (miRNAs), has been extensively attracted in the field of HD. Previously, we found miR-196a improves neuropathological and behavioral phenotypes in HD [4, 5], suggesting that miRNA-mediated pathways should contribute to this neurodegenerative disease, and that alteration of miRNAs might provide neuroprotective effects in HD.
Neuroprotective effects can function in the brain through improvements with regard to neurogenesis, anti-apoptosis, anti-inflammation, neuronal morphology and mitochondria functions. In particular, neuronal morphology is one of the most important effects, as this influences intracellular trafficking, synaptic development, plasticity and brain circuitry, further affecting motor, memory and cognitive functions in the brain [6-9]. In addition, neuronal morphology is also highly regulated by miRNAs, leading to the establishment of functional neurons [10, 11]. These results suggest neuroprotection might be regulated by miRNAs through enhancement of neuronal morphology.
In our previous study, we found one specific miRNA, miR-196a, provides beneficial effects on HD via cell, transgenic mouse and HD patient-derived induced pluripotent stem cell (HD-iPSC) models [4]. Moreover, we further showed that miR-196a putatively provides beneficial functions through the alteration of brain cell cytoskeleton structures in HD [5]. In this study, we investigated the roles of miR-196a in neuronal morphology, and demonstrated the regulatory mechanisms through modification of the cytoskeleton structures. Furthermore, we also addressed the related mechanisms in HD, and anticipate that the results of this work will provide some directions for the development of novel therapeutic strategies.
Materials and methods
DNA constructions
The DNA transgenes used in this study include Ds-Red, miR-196a-DsRed, RANBP10, RANBP10-dsRed, shRANBP10, G19Q and G84Q. Ds-Red contains a red fluorescence protein (RFP) gene under control of a human ubiquitin promoter. miR-196a-DsRed carries the precursor hsa-miR-196a-2 (accession number: MI0000279) under control of a human ubiquitin promoter and a RFP gene for observation. RANBP10 contains RAN Binding Protein 10 gene (accession number: NM_145824) under control of a human ubiquitin promoter. RANBP10-dsRed carries RANBP10 under control of a human ubiquitin promoter and a RFP gene for observation. shRANBP10 contains CCGGCCAGTAGGTCATCAGCTTGATCTCGAGATCAAGCTGATGACCTACTGGTTTTTG in a pLKO.1 vector. G19Q and G84Q contains a green fluorescence protein gene fused with the exon 1 region of the Huntingtin gene carrying 19 and 84 CAG repeats, respectively, under control of a human ubiquitin promoter. All these transgenes are inserted into a lentiviral vector (a gift from Dr. David Baltimore; Addgene plasmid: #14883). In addition, a RANBP10 3'UTR construct is used for a luciferase reporter assay. The RANBP10 3'UTR construct contains 3' untranslated region (3'UTR) of RANBP10 (accession number: NM_145824) flanking to the 3' end of a luciferase gene in a PIS2 vector (Addgene plasmid: #12177).
Primary cortical neurons
The animal protocol performed in this study was approved by the Institutional Animal Care and Use Committee at National Cheng Kung University, Taiwan. FVB mouse embryos were collected at 17.5 days post coitum (dpc), and primary neurons were isolated from the cortex tissues of these embryos and cultured in Neurobasal Medium (Gibco) supplemented with 1X B27 (Gibco) and 2mM L-glutamine (Gibco) in an incubator. To perform transfection, primary cortical neurons were cultured for three days, and then transfected with foreign transgenes using lipofectamineTM 2000 (Invitrogen). Transfected cells were kept culturing for four days, and neuronal morphology was evaluated.
Neurite outgrowth
Evaluation of neurite outgrowth was performed in N2a mouse neuroblastoma cells and primary neurons according to the previous study [5]. In N2a cells, cells were transfected with different transgenes, and differentiated via culture medium with 10 μM retinoic acid (Sigma) and 2% fetal bovine serum (Hyclone) for 48 hours. In primary cortical neurons, the transfected cells described above were used for analyses. The images of neurite outgrowth were captured under a DM2500 fluorescent microscope (Leica) and analyzed though the Neurite Outgrowth Application Module of the MetaMorph software (Molecular Devices).
Golgi stain
An FD Rapid GolgiStainTM Kit (FD Neurotechnologies, Inc) was used for Golgi staining following the manufacturer's protocol. In brief, brains were stained using Solution A and Solution B, transferred to Solution C, and then subjected for cryosectioning using a cryostat (Thermo). Slides with a thickness of 140 μm were stained using the mix of Solution D and E, and fixed for analyzing neurite outgrowth. Neuronal morphology was captured every 2 μm along the z axis from these stained slides using a DMi8 microscope (Leica), and then images were staged into two-dimensional images using MetaMorph software (Molecular Devices). The staged images were quantitated by NeuronJ, which is an ImageJ plugin (NIH), and then subjected to statistical analyses.
Intracellular transport
The examination of intracellular transport was modified and conducted according to a previous study [12]. N2a cells were contransfected by Ds-Red, miR-196a-DsRed or RANBP10-dsRed with the amyloid precursor protein-yellow fluorescent protein (APP-YFP) fusion gene for 18 hours. Cells were then cultured on a heating stage (Live Cell Instrument) with a DMi8 microscope (Leica), and fluorescent images were recorded every 0.44 seconds for 66 seconds via the control of MetaMorph, which was also used to analyze the moving distance and velocity of the APP-YFP fusion protein.
Microtubule polymerization
Microtubule depolymerization and repolymerization were determined according to the method developed in a previous study [13]. N2a cells were transfected with Ds-Red, miR-196a-DsRed or RANBP10-dsRed, cultured at 37℃ for 48 hours, and some cells were fixed using 4% paraformaldehyde as the control stage. Other transfected N2a cells were kept incubated on ice (4℃) for 15 minutes, and then fixed as the depolymerization stage. Some cells were kept culturing at 37℃ for 24 hours, and then fixed at the repolymerization stage. These fixed cells at different stages were subjected to immunofluorescent staining using a β-tubulin antibody (GenTex, GTX101279), and total neurite outgrowth was determined using MetaMorph.
Luciferase reporter assay
A RANBP10 3'UTR construct in a PIS2 vector was used for luciferase reporter assay as described in DNA constructs above. Site-directed mutagenesis of the miR-196a binding site in RANBP10 3'UTR was generated using PCR amplification as shown in Figure 2a. 293 FT cells were co-transfected with RANBP10 3'UTR/ a cytomegalovirus-driven β-gal reporter system/ miR-196a mimics or non-relative controls using lipofectamineTM 2000 (Invitrogen) for 24 hours, and cell pellets were subjected to a Luciferase reporter assay according to the manufacturer's instructions (Progema). β-gal reporter system was used as an internal control. The miR-196a mimics used in this study were annealed by 5'-UAGGUAGUUUCAUGUUGUUGGG-3' and 5'-CAACAACAUGAAACUACCUAUU-3', and non-relative controls were annealed by 5'-UUCUCCGAACGUGUCACGUTT-3' and 5'-ACGUGACACGUUCGGAGAATT-3' (MD Bio, Inc.). The luciferase activities in different groups were quantitated and subjected to statistical analyses.
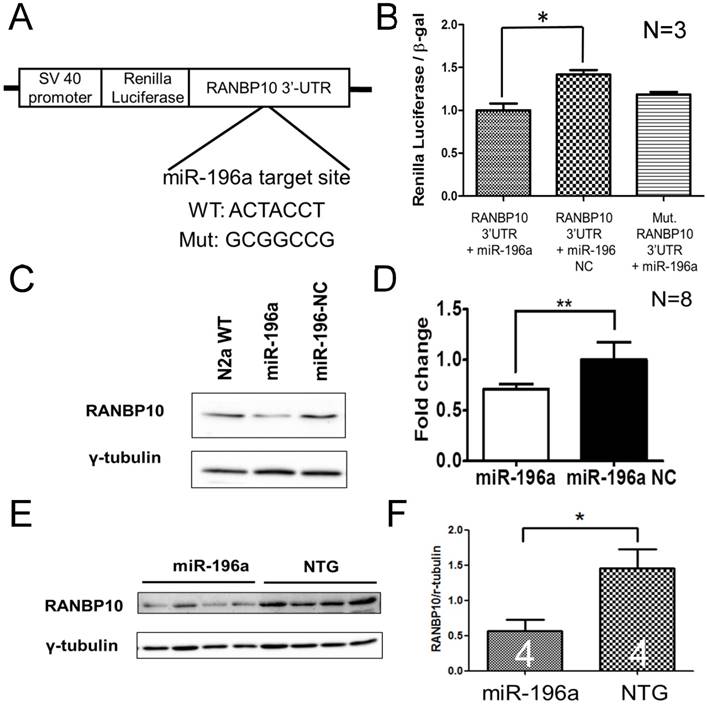
miR-196a suppresses the expression of RANBP10 through binding to 3'UTR of RANBP10. N2a cells were co-transfected with miR-196a and a reporter construct to determine the direct binding of miR-196a on 3'UTR of RANBP10. N2a cells transfected with miR-196a and the brains of miR-196a transgenic mice were used to determine the suppression of endogenous RANBP10. (A) Wild-type or mutant (mut.) 3'UTRs of RANBP10 was constructed into the 3' end of luciferase gene for the reporter assay. (B) Luciferase reporter assay shows miR-196a binds to 3'UTR of RANBP10 to suppress the expression of luciferase activity compared to those in the miR-196a non-relative control (NC) or mutant 3'UTR of RANBP10 (Mut. RANBP10 3'UTR). N=3. (C) Western blotting shows the expression of RANBP10 after treatment of miR-196a mimics and NC in N2a cells. (D) Quantitation results show the suppression of RANBP10 after treatment of miR-196a mimics in N2a cells. N=8. (E) Western blotting shows the expression of RANBP10 in the brains of miR-196a transgenic and non-transgenic (NTG) mice. (F) Quantitation results show the lower expression of RANBP10 in miR-196a transgenic mice. *indicates a significant difference with P<0.05. **indicates a significant difference with P<0.01.
Western blotting analysis
Cell and brain samples were lysed using a sonicator (Qsonica), and were subjected to Sodium dodecyl sulfate polyacrylamide gel electrophoresis (Bio-Rad). Separated proteins were transferred onto PVDF membranes (Bio-Rad) for Western blotting, and then hybridized with the primary antibodies, including P-ERK (Cell Signaling; 1:1,000 dilution), T-ERK (Cell Signaling; 1:2,000 dilution), P-GAP43 (Abcam; 1:1,000 dilution), T-GAP43 (Genetex; 1:5,000 dilution), PSD95 (Abcam; 1:5,000 dilution), Synaptophysin (Abcam; 1:5,000 dilution), VAMP1 (Abcam; 1:5,000 dilution), RANBP10 (Proteintech; 1:2,000), Flag M5 (Sigma; 1:2,000 dilution), mEM48 (a gift from Dr. Xiao-Jiang Li, Emory University, USA; 1:50 dilution), MAB2166 (Millipore; 1:2,000 dilution) and γ-tubulin (Sigma; 1:10,000 dilution) antibodies. Protein expression levels were determined by an Amersham ECL kit (PerkinElmer) through an imaging system (ChampGel), and expression profiling was quantitated by an ImageJ system (NIH).
Immunohistochemical staining
Transfected cells were fixed, blocked and hybridized with primary antibodies, including P-GAP43 (Abcam; 1:500 dilution), RANBP10 (Proteintech; 1:500) and β-tubulin (Sigma; 1:1,000 dilution) antibodies. The cells were then reacted with secondary antibodies conjugated with Alexa fluor 488, 594 or 647 (Invitrogen), and cellular nuclear were stained by 1μg/mL Hoechst 33342 (Sigma). Fluorescence images were captured using a DM2500 fluorescent microscope (Leica) for analyses. For 3,3'-Diaminobenzidine (DAB) immunohistochemical staining, brain sections were blocked, incubated with c-Fos (Cell signaling; 1:250) and examined with a Vectastain Elite ABC kit (Vector Laboratories) and DAB (Vector Laboratories). The intensity of c-Fos was quantitated using ImageJ (NIH), and results were subjected to statistical analyses.
Electrophysiology
Mouse brains were subjected for horizontal sectioning, and subjected to extracellular recordings of field excitatory postsynaptic potentials (fEPSPs) using a recording electrode in the CA1 region of the hippocampus and a stimulating electrode in the CA3 region. The baseline of fEPSP was monitored, and then high-frequency stimulation (HFS) at 100Hz was applied. Evoked fEPSPs were recorded via an AxoClamp-2B amplifier (Molecular Devices), and analyzed using pClamp 10.5 software (Molecular Devices). The long-term potentiation (LTP) of CA1 was calculated via the averaged changes of fEPSP slopes at 0 and 40 minutes after HFS.
Transgenic mice
The transgenic mice used in this study include miR-196a, RANBP10, R6/2 and RANBP10-R6/2. The animal protocol performed in this study was approved by the Institutional Animal Care and Use Committee at National Cheng Kung University, Taiwan. MiR-196a transgenic mice carry the precursor hsa-miR-196a-2 (accession number: MI0000279) under control of a human ubiquitin promoter, as described before [4, 5]. RANBP10 transgenic mice carry RANBP10 gene (accession number: NM_145824) under control of a human ubiquitin promoter. R6/2 mice are HD transgenic mice carrying truncated exon1 with expanded polyQ under control of a HTT promoter [14]. The RANBP10-R6/2 transgenic mice were double transgenic mice (D-Tg) obtained through breeding RANBP10 transgenic mice with R6/2 transgenic mice.
Behavioral examinations
T-maze test
A T-maze with three arms, including two goal-arms and one stem-arm plumb to goal-arms, was used in this study. Mice were fasted for 18 hours before the tests, and training and testing parts were conducted. During the training period, food with condensed milk was placed in one goal-arm, and one mouse was trained to memorize the location of the goal-arm with food for four minutes, three times, with one-minute intervals. During the testing period, food was removed completely, and one mouse was placed into the T-maze for four minutes. The movement of this mouse was monitored by video-recording, and the resulting videos were examined using TopScan LITE software (Clever Sys Inc.) to analyze the time spent in the two different goal-arms.
Novel object recognition test
A hollow box was used, and the test includes training and testing parts. During the training part, two identical objects were set in the field and one mouse was trained to memorize these for 4 minutes, three times, with one-minute intervals. During the testing period, one of the objects was replaced by a new and different item, and the trained mouse was then placed into the same field for four minutes. The movement of this mouse was monitored by video-recording, and the videos were examined using TopScan LITE to analyze the time spent with the two different objects.
Statistical analyses
Data are presented as the mean ± standard error of mean (SEM). Student's t-test was used to compare differences between two different groups. On some occasions, differences among different groups were analyzed using one-way analysis of variance using commercial statistical software (GraphPad Prism 4.02; GraphPad Software, San Diego, CA). Tukey's procedure was used to test differences among different groups. Statistical significance was set at P < 0.05.
Results
Our previous studies showed that miR-196a improves the molecular, neuropathological and behavioral phenotypes of HD in different models [4] and miR-196a might putatively regulate the cytoskeletons of brain cells in HD [5]. Here, we attempt to further demonstrate the neuroprotective functions of miR-196a through neuronal morphology in HD.
First, we examine whether miR-196a could enhance neurite outgrowth in primary neurons in vitro or transgenic mice in vivo. We determined the branches and total neurite length in the transfected primary neurons (Supplementary Figure 1A and 1B), showing significantly more branches and longer neurite length in miR-196a neurons (Figure 1A). Furthermore, the neurons in the frontal cortex of miR-196a transgenic mice displayed significantly more branches and longer neurite length compared to those of non-transgenic mice (Supplementary Figure 1C and 1D; Figure 1B). These results suggest that miR-196a could enhance the neuronal differentiation and morphology in vitro and in vivo.
miR-196a enhances neuronal morphology, intracellular transport, synaptic plasticity, neuronal activity and learning and memory. (A) Primary cortical neurons were transfected with Ds-Red or miR-196a-DsRed, and Quantitation results show increased branches (left) and total neurite length (right) after miR-196a treatment. Primary neurons from three batches were examined. (B) The brains of non-transgenic (NTG) and miR-196a transgenic mice were subjected to Golgi staining, and quantitation results show the increased branches (left) and total neurite length (right) in the brains of miR-196a transgenic mice. Three mice from each group were examined. The number of examined neurons is indicated. (C) N2a cells cotransfected with Ds-Red & APP-YFP or miR-196a-Ds-Red & APP-YFP were used for examining intracellular transport, and quantitated results show the significantly faster velocity of APP-YFP movement during anterograde but not retrograde transport in miR-196a-Ds-Red cells. The number of examined neurons is indicated. (D-J) Brains of miR-196a transgenic mice were used for Western blotting, immunohistochemical staining and electrophysiological examination. Non-transgenic (NTG) and miR-196a transgenic mice were subjected to behavioral tests. (D) Western blotting results show the expression levels of PSD95, Synaptophysin and VAMP1 in miR-196a transgenic and NTG mice. γ-tubulin was used as an internal control. (E) Quantitation results show the increase of VAMP1/γ-tubulin in miR-196a transgenic mice. (F) Immunohistochemical staining shows the expression profiling of c-Fos in hippocampal regions of miR-196a transgenic and NTG mice. The top panel shows lower magnification images, and the bottom panel shows higher magnification of CA2-CA3 regions. (G) Quantitation results show the significant increase in c-Fos+ cells, which carry a higher intensity of c-Fos signal, in miR-196a transgenic mice. (H) Western blotting shows the expression of c-Fos in the brains of wild-type (WT) and miR-196a transgenic mice. γ-tubulin was used as an internal control. (I) Quantitation results from (H) show the significant increase of c-Fos/γ-tubulin expression in miR-196a transgenic mice. (J) fEPSP in CA1 regions was determined at different time points, and the long-term potentiation (LTP) at 40 minutes after high-frequency stimulation (HFS) is also shown. The top two graphs show the representative fEPSP. Black lines indicate traces before HFS, and red lines indicate traces at 40 minutes after HFS. (K) Quantitation results of the T-maze test show a significant difference between the two groups. * indicates a significant difference with P<0.05; ** indicates a significant difference with P <0.01. ***indicates a significant difference with P<0.001.
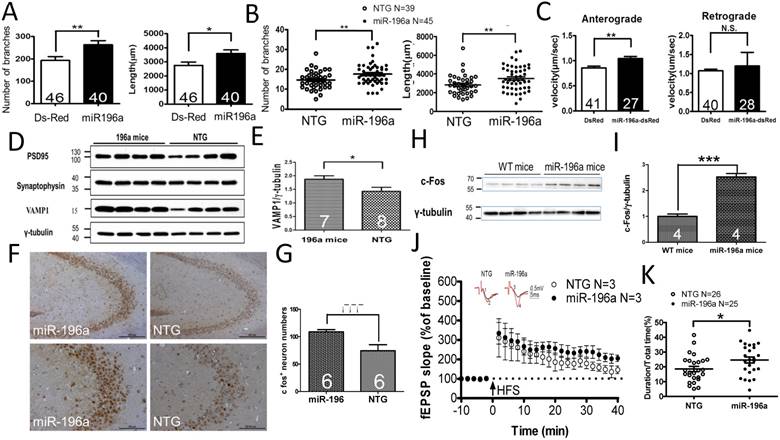
Several cellular signals have been reported to be involved in the regulation of neuronal morphology, such as extracellular signal-regulated kinase (ERK) and Growth Associated Protein 43(GAP-43) [15]. We examined the expression profiling of two critical proteins, ERK and GAP-43, in miR-196a transgenic mice, and the expression levels were significantly increased in miR-196a transgenic mice (Supplementary Figure 2A and 2B), suggesting miR-196a could enhance neuronal morphology via increases in ERK and GAP43 signaling.
Neuronal morphology plays an important role in intracellular transport, and well-established neurites provide better effects with regard to this function [16]. Here, amyloid precursor protein-yellow fluorescent protein (APP-YFP) was used as a reporter cargo for recording intracellular transport [12]. We confirmed the expression of APP-YFP with DsRed or miR-196a DsRed in N2a cells (Supplementary Figure 3A), and recorded the serial live images for intracellular transport of APP-YFP (Supplementary Figure 3B). As shown in Figure 1C, we observed increased velocity of intracellular transport during anterograde but not retrograde transport in N2a cells overexpressing miR-196a compared to that of DsRed control, suggesting miR-196a could enhance not only neuronal morphology but also intracellular transport.
Intracellular transport is a critical factor affecting synaptic plasticity and neuronal activity in neuronal cells [17]. Therefore, we further investigated the effects of miR-196a on synaptic plasticity/functions, First, we determined the expression of synaptic proteins in transgenic mice, showing that the expression of PSD95 and Synaptophysin between miR-196a and non-transgenic mice was not significantly different, but VAMP1 was significantly increased in miR-196a transgenic mice (Figure 1D and 1E). Additionally, we also determined neuronal activity in the brains of these transgenic mice via the detection of c-Fos [18], showing miR-196a transgenic mice had increased intensity of c-Fos signals in the brains using immunohistochemical staining and Western blotting (Figure 1F, 1G, 1H and 1I). We also examined the expression profiling of CalbindinD-28k, a marker related to neuronal activity and synaptic plasticity [19], and found higher expression levels of CalbindinD-28k in miR-196a transgenic mice (Supplementary Figure 4). Most importantly, electrophysiology was applied to determine the long-term potentiation (LTP) of CA1 in these mice, and we did observe the trend of stronger LTP in miR-196a transgenic mice (P=0.09; Figure 1J). Furthermore, because neuronal activity and synaptic plasticity are highly related to learning and memory, we performed T-maze and novel object recognition tests to examine the ability of learning and memory in these mice. As a result, miR-196a transgenic mice showed significantly better abilities of learning and memory in the T-maze test (Figure 1K), but not in the novel object recognition test (Supplementary Figure 5). Taken together, these results suggest that miR-196a could enhance neuronal morphology, intracellular transport, neuronal activity, synaptic plasticity and learning and memory ability in transgenic mice in vivo.
Since miRNAs predominantly function through binding to the 3' untranslated region (3'UTR) of target genes to process the miRNA machinery, we are more interested in identifying the direct targets of miR-196a that are involved in above functions of morphological alterations. To achieve this, we used an on-line tool, TargetScan (http://www.targetscan.org/), to predict one specific target gene, Ran-binding protein 10 (RANBP10), because this has been shown to regulate microtubule equilibrium in platelets [13, 20]. In order to confirm whether miR-196a could bind to 3'UTR of the RANBP10 gene, we cloned wild-type and mutant 3'UTR of RANBP10 flanking to the 3' end of the luciferase gene to form a reporter construct (Figure 2A), and observed that miR-196a could bind to 3'UTR of RANBP10 to significantly suppress the expression of luciferase activity in the wild-type group, not in mutant one (Figure 2B). We further examined the expression level of endogenous RANBP10 in vitro and in vivo after overexpression of miR-196a, showing a significant decrease of endogenous RANBP10 in miR-196a transfected cells (Figure 2C and 2D) and miR-196a transgenic mice (Figure 2E and 2F). These results suggest miR-196a could bind to 3'UTR of RANBP10 to suppress the expression of RANBP10.
To examine whether miR-196a functions on the above phenotypes through RANBP10, an overexpression construct of RANBP10 under control of an ubiquitin promoter was generated and confirmed (Supplementary Figure 6A and 6B). Additionally, RANBP10 transgenic mice were generated for in vivo studies (Supplementary Figure 6C) [21]. Since miR-196a suppresses the expression of RANBP10 and also provides beneficial functions in the above studies, we hypothesize that RANBP10 might exacerbate neuronal morphology and functions. Conducting similar examinations, overexpression of RANBP10-DsRed resulted in significantly less branches and shorter neurite outgrowth in N2a (Supplementary Figure 7A and 7B) and primary neurons (Supplementary Figure 7C and 7D). Furthermore, RANBP10 transgenic mice showed the decreased total neurite length (Supplementary Figure 7E and 7F). Moreover, RANBP10 significantly decreased the transport velocity of APP-YFP during retrograde but not anterograde transport (Supplementary Figure 8). These results suggest RANBP10 did exacerbate neuronal morphology and intracellular transport.
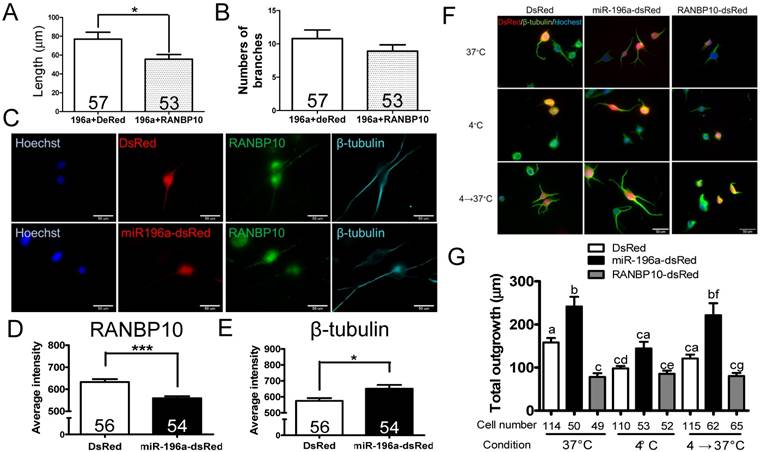
Since miR-196a enhances neuronal morphology, miR-196a directly targets to RANBP10 and RANBP10 worsens neuronal morphology, we speculate whether overexpression of RANBP10 leads to blockage of miR-196a functions on neuronal morphology. We contransfected miR-196a-GFP with RANBP10-dsRed into N2a cells, and observed neurite outgrowth, showing RANBP10 significantly blocks the function of miR-196a on total neurite length (Figure 3A), but not on branch numbers (Figure 3B). This suggests RANBP10 is a critical regulator of miR-196a on neuronal morphology. Furthermore, based on previous studies RANBP10 has been shown to regulate β-tubulin equilibrium in platelets [13, 20], suggesting miR-196a might suppress RANBP10 to enhance cellular morphology through improving the organization of β-tubulin. We thus co-immunostained endogenous RANBP10 and β-tubulin in N2a cells transfected with DsRed or miR-196a-dsRed, and showed that a decreased intensity of RANBP10 and increased intensity of β-tubulin were observed in cells transfected with miR-196a-dsRed compared with the results found for the non-transfected ones (Figure 3C, 3D and 3E), suggesting that miR-196a suppresses RANBP10 to enhance cellular morphology through better organization of β-tubulin. Furthermore, the good organization of β-tubulin might due to its stabilization or the enhancement of β-tubulin polymerization. To examine this issue, N2a cells transfected with DsRed, miR-196a-dsRed or RANBP10-DsRed were serially cultured at different temperatures, and then fixed and stained using a β-tubulin antibody to determine β-tubulin organization (Figure 3F). Similar to the earlier studies noted above, better β-tubulin polymerization in miR-196a-dsRed cells and worse β-tubulin polymerization in RANBP10-DsRed cells at 37℃ were observed compared to that seen in DsRed cells (Figure 3F top panel; 3G). When treated at 4℃ to disrupt the β-tubulin polymerization, less total neurite outgrowth was observed, except in the RANBP10-DsRed cells (Figure 3F, middle panel; 3G). Moreover, the cells were re-cultured at 37℃ to examine the ability of repolymerization, and only miR-196a-dsRed cells were significantly better in this regard (Figure 3F, bottom panel; 3G). These results suggest that miR-196a enhances neuronal morphology through increasing the ability of β-tubulin polymerization.
miR-196a enhances neuronal morphology through suppressing RANBP10 to enhance β-tubulin polymerization. N2a cells were transfected 196a-GFP and RANBP10-dsRed to determine neurite outgrowth in (A) and (B). N2a cells contransfected with DsRed, miR-196a-dsRed or RANBP10-dsRed, cultured at different temperature conditions and subjected to immunostaining using RANBP10 and β-tubulin antibodies in (C)-(G). (A) Overexpression of RANBP10 blocks the function of miR-196a on total neurite outgrowth, not number of branches in N2a cells (B). (C) Representative images show the expression profiles of RANBP10 and β-tubulin in N2a cells transfected with DsRed (top panel) or miR-196a-dsRed (bottom panel). Hoechst (blue) staining shows the location of the nuclei. (D) Quantitative results show the significantly lower expression level of RANBP10 in miR-196a treatment cells. (E) Quantitative results show the significantly higher expression level of β-tubulin in miR-196a treatment cells. (F) Immunostaining results show the expression profiles of β-tubulin (green) in N2a cells transfected with DsRed (left panel), miR-196a-dsRed (middle panel) or RANBP10-dsRed (right panel) at different temperature conditions. Hoechst staining (blue) shows the location of nuclei, and transfected cells are indicated by red fluorescence. (G) Quantitative results show miR-196a significantly enhances repolymerization of β-tubulin when cultured from 37℃ to 4℃, whereas RANBP10 disrupts the neuronal morphology at different temperatures. Three batches of experiments were performed in this study. * indicates a significant difference with P <0.05. *** indicates a significant difference with P <0.001. Different letters on different bars in (e) indicate a significant difference with P <0.05.
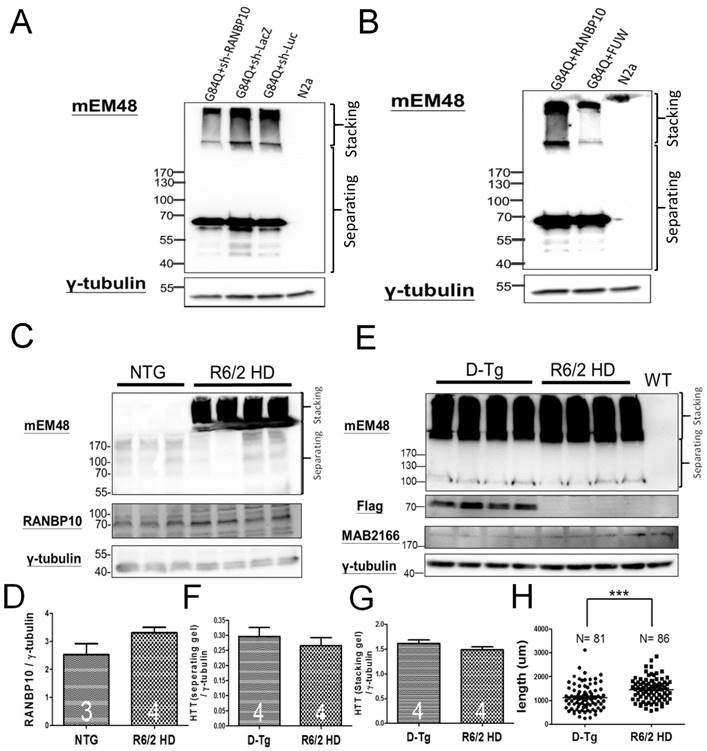
Finally, since we previously reported the protective effects of miR-196a in different HD models [4], we are also interested in the role of RANBP10 in HD. To examine this question, we first attempted to alter the expression of RANBP10 in N2a cells transfected with mutant HTT, and detected the aggregation of mutant HTT via Western blotting. We confirmed the functions of RANBP10 or RANBP10 short hairpin RNA (shRANBP10) constructs in vitro (Supplementary Figures 6 and 9), and found that less aggregated HTT was observed at a high molecular weight in stacking gel when RANBP10 was knocked down, whereas more aggregated HTT was found stacked at a high molecular weight when RANBP10 was overexpressed (Figure 4A and 4B; Supplementary Figure 10). In addition, we wonder whether overexpression of RANBP10 also exacerbates pathological aggregates using HTT with normal CAG repeats. We overexpressed RANBP10 and HTT with 19 CAG repeats (G19Q), and found RANBP10 does not lead to aggregates and abnormal expression of HTT (Supplementary Figure 10). In in vivo research, the expression profiling of RANBP10 was examined in R6/2 HD transgenic mice [14], showing a greater trend of RANBP10 expression in R6/2 transgenic mice (Figure 4C and 4D). To further confirm the harmful effects of RANBP10 in HD, we detected the aggregate profiling in brains of R6/2-RANBP10 double transgenic mice (D-Tg) overexpressing mutant HTT and RANBP10, and aggregates of mutant HTT were displayed in both D-Tg and R6/2 mice (Figure 4E). Further quantitating the aggregates in separating and stacking gels, although there was no significant difference between these two groups, higher trends of mutant HTT expression were measured in D-Tg compared to those seen in R6/2 mice (Figure 4F and 4G). In addition, we determined the effects of RANBP10 on endogenous HTT in vivo using a MAB2166 antibody, which only recognizes the mouse endogenous HTT in this study, and we found the expression level of endogenous mouse HTT was not affected by the overexpression of RANBP10 in brains of transgenic mice (Figure 4e and Supplementary Figure 11), suggesting overexpression of RANBP10 does not influence the expression of endogenous HTT in vivo. Moreover, the neurons in the frontal cortex of D-Tg transgenic mice displayed significantly shorter neurite length compared to those of R6/2 mice (Figure 4H). Taken together, these results suggest that RANBP10 might exacerbate the pathological aggregates in HD.
RANBP10 exacerbates pathological aggregates in vitro and in vivo in HD. For the in vitro study, N2a cells were cotransfected G84Q with shRANBP10 or RANBP10 for 48 hours, and cell samples were collected for Western blotting (A and B). For the in vivo study, the brains of non-transgenic (NTG), R6/2 HD or D-Tg transgenic mice at 1.5 months of age were subjected to Western blotting. (A) Western blotting shows the expression of mutant HTT using a mEM48 antibody after cotransfection of G84Q with shRANBP10(suppression), sh-lacZ (negative control) or sh-luc (negative control). γ-tubulin was used as an internal control. (B) Western blotting shows the expression of mutant HTT using a mEM48 antibody after cotransfection of G84Q with RANBP10 (overexpression) and FUW (empty vector control). γ-tubulin was used as an internal control. (C) Western blotting shows the expression of mutant HTT in brain tissues of NTG and R6/2 mice. mEM48 and RANBP10 antibodies were used to detect mutant HTT and endogenous RANBP10, respectively. (D) Quantitation results show lower expression of endogenous RANBP10 in NTG mice compared to those of R6/2 mice. (E) Western blotting shows the expression of mutant HTT in brain tissues of D-Tg and R6/2 mice. mEM48, Flag and MAB2166 antibodies were used to detect mutant HTT, exogenous RANBP10 and endogenous mouse HTT, respectively. (F) Quantitation results show higher expression of mutant HTT in separating gel in D-Tg compared to those of R6/2 mice. (G) Quantitation results show higher expression of mutant HTT in stacking gel in D-Tg compared to those of R6/2 mice. The number of examined mice is indicated inside different bars. (H) The brains of D-Tg and R6/2 mice were subjected to Golgi staining, and quantitation results show the decreased total neurite length in the brains of D-Tg transgenic mice. Three mice from each group were examined. The number of examined neurons is indicated. *** indicates a significant difference with P <0.001.
Discussion
To date there is no cure for neurodegenerative diseases, such as Huntington's disease, and it is important that work continues with regard to developing therapeutic treatments for these diseases. miR-196a has been shown to be a potential target to act against Huntington's disease [4, 5], although the working mechanisms remain unclear. In this study, we show the miR-196 could enhance neuronal morphology, intracellular transport, synaptic plasticity and learning and memory. In addition, miR-196a could suppress RANBP10 to improve neuronal morphology through increasing the ability of β-tubulin polymerization. Furthermore, overexpression of RANBP10 harmed neuronal morphology and functions, even further leading to greater HTT aggregates. These results not only show the effects of miR-196a on neuronal morphology and the resulting phenotypes, but also show one neuroprotective mechanism of miR-196a in HD.
miR-196a, a highly conserved miRNA, is located in the mammalian HOX clusters, and has been reported to be involved in the development of embryos and different organs [22-25]. Moreover, miR-196a is highly involved in cancer development, although both positive and negative effects have been shown for cancer progression [26-28]. These findings suggest that miR-196a plays an important role during the development and progression of various diseases. As a result, the side effects of the potential application of miR-196a are also considered. Based on previous studies, miR-196a could alleviate the progression of spinal and bulbar muscular atrophy and Huntington's diseases without any side effects being reported [4, 29]. In our miR-196a transgenic mice, normal life span and healthy appearance were observed, thus providing further evidence that miR-196a could have potential therapeutic applications.
RANBP10 is a cytoplasmic guanine nucleotide exchange factor (GEF), and binds to β-tubulin to provide GEF activity toward RAN, which regulates microtubule spindle assembly during mitosis [30]. In addition, RANBP10 is involved in microtubule equilibrium to maintain platelet shape [13, 20]. Our data also showed higher expression of RANBP10 worsens the neurite outgrowth (Supplementary Figure 7), further supporting the role of RANBP10 in the organization of the cytoskeleton and resulting functions, such as intracellular transport (Supplementary Figure 8). More interestingly, our male RANBP10 transgenic mice displayed a lower germline transmission rate (less than 5%) when we bred these transgenic mice. This implies that RANBP10 might disrupt the cytoskeleton of sperm, leading to a worse swimming ability of those carrying RANBP10. However, further investigation is still needed to demonstrate reproductive problems in these transgenic mice.
Furthermore, RANBP10 functions as a coactivator for the androgen receptor and several viral genes [31, 32], so we speculate that miR-196a might act through not only the cytoskeleton, but also gene regulation. Most interestingly, the fact that spinal and bulbar muscular atrophy (SBMA) is also a polyglutamine disease, caused by expansion of CAG repeats in the androgen receptor gene, and miR-196a can ameliorate the SBMA phenotypes [29], is highly suggestive of the important role of RANBP10 in polyglutamine diseases, including Huntington's disease.
Moreover, another issue was raised when we detect the expression of RANBP10. We detected RANBP10 at approximately 55kD in N2a cells and mouse tails (Figure 2C and supplementary Figure 7C), but detected at approximately 70kD in other results of RANBP10. We were confused by this result. Based on the cDNA sequence we cloned, we expected the molecular weight of RANBP10 is approximately 70kD. Therefore, we subjected the 55kD band to the analysis of mass spectrometry, and found the protein sequence did match to RANBP10 (Supplementary Figure 12). Therefore, although we do not exactly know why to form two different bands, we believe these bands are RANBP10.
With regard to intracellular transport, increased velocity during anterograde transport was observed when cells were treated with miR-196a (Figure 1C; Supplementary Figure 3). In contrast, the decreased velocity during retrograde transport, but not anterograde, was shown in cells treated with RANBP10 (Supplementary Figure 8). These results suggest that miR-196a might not only accelerate anterograde transport, but also suppress the deficit in retrograde transport during intracellular transport. In addition, these results also imply that miR-196a might function through RANBP10 and other regulatory pathways during trafficking. As a result, different effects on anterograde and retrograde transport were observed when miR-196a or RANBP10 was overexpressed. Since the working mechanism of miRNAs is usually through several target genes, this suggests that other target genes might also be involved in intracellular transport. In particular, the neuroprotective effects of miR-196a were originally reported in HD [4], and a deficit in neuronal transport is a critical phenotype of HD [33]. It would thus be interesting to examine the miR-196a target genes related to the regulation of kinesin and dynein, which are two critical proteins responsible for anterograde and retrograde transport, respectively [34].
Abnormal neuronal morphology has been reported in HD [5, 35]. In addition, miR-196a has been shown to improve the phenotypes of this disease [4, 36]. In this study, a higher expression of RANBP10 not only showed exacerbation of neuronal morphology and related functions, but also led to more pathological aggregates in HD models. Since miR-196a has also shown beneficial functions with regard to other neuronal diseases [29] and miR-196a is anticipated to inhibit the expression of RANBP10 in different neuronal cells, this strongly indicates that enhancement of neuronal morphology is one of the neuroprotective functions provided by miR-196a through suppression of RANBP10 in different neuronal diseases. However, the neuroprotective roles of miR-196a in other neuronal diseases still require further examination.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Paul Steed for careful reading and editing, Professor Fong-Sen Wu's support for brain electrophysiology, and all members in Yang's laboratory at National Cheng Kung University. This work was supported by the Ministry of Science and Technology (MOST 105-2628-B-006-015-MY3, MOST 104-2320-B-006 -044 and MOST 105-2320-B-006-004) and, in part, the Ministry of Education, Taiwan, R.O.C., under The Aim for the Top University Project to the National Cheng Kung University (NCKU).
Authors' contributions
LSH, SHM, CYC, PHC, YFC and SHY handled animal studies, molecular analysis and analyzed data. LSH, CMC and SHY designed the experiments and oversaw the progression of this study. LSH, CMC and SHY drafted the paper. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Group THsDCR. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971-83
2. Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR. et al. Huntington disease. Nat Rev Dis Primers. 2015:15005
3. Yang SH, Chan AW. Transgenic Animal Models of Huntington's Disease. Curr Top Behav Neurosci. 2011;7:61-85
4. Cheng PH, Li CL, Chang YF, Tsai SJ, Lai YY, Chan AW. et al. miR-196a Ameliorates Phenotypes of Huntington Disease in Cell, Transgenic Mouse, and Induced Pluripotent Stem Cell Models. Am J Hum Genet. 2013
5. Fu MH, Li CL, Lin HL, Tsai SJ, Lai YY, Chang YF. et al. The Potential Regulatory Mechanisms of miR-196a in Huntington's Disease through Bioinformatic Analyses. PloS one. 2015;10:e0137637
6. Olde Loohuis NF, Kos A, Martens GJ, Van Bokhoven H, Nadif Kasri N, Aschrafi A. MicroRNA networks direct neuronal development and plasticity. Cellular and molecular life sciences. Cell Mol Life Sci. 2012;69:89-102
7. Skaper SD. Neuronal growth-promoting and inhibitory cues in neuroprotection and neuroregeneration. Ann N Y Acad Sci. 2005;1053:376-85
8. Calabrese EJ. Enhancing and regulating neurite outgrowth. Crit Rev Toxicol. 2008;38:391-418
9. Bencsik N, Sziber Z, Liliom H, Tarnok K, Borbely S, Gulyas M. et al. Protein kinase D promotes plasticity-induced F-actin stabilization in dendritic spines and regulates memory formation. J Cell Biol. 2015;210:771-83
10. Hennchen M, Stubbusch J, Abarchan-El Makhfi I, Kramer M, Deller T, Pierre-Eugene C. et al. Lin28B and Let-7 in the Control of Sympathetic Neurogenesis and Neuroblastoma Development. J Neurosci. 2015;35:16531-44
11. Franzoni E, Booker SA, Parthasarathy S, Rehfeld F, Grosser S, Srivatsa S. et al. miR-128 regulates neuronal migration, outgrowth and intrinsic excitability via the intellectual disability gene Phf6. eLife. 2015:4
12. Her LS, Goldstein LS. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 2008;28:13662-72
13. Meyer I, Kunert S, Schwiebert S, Hagedorn I, Italiano JE Jr, Dutting S. et al. Altered microtubule equilibrium and impaired thrombus stability in mice lacking RanBP10. Blood. 2012;120:3594-602
14. Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C. et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493-506
15. Neumann JR, Dash-Wagh S, Jungling K, Tsai T, Meschkat M, Rak A. et al. Y-P30 promotes axonal growth by stabilizing growth cones. Brain Struct Funct. 2015;220:1935-50
16. Kapitein LC, Hoogenraad CC. Building the Neuronal Microtubule Cytoskeleton. Neuron. 2015;87:492-506
17. Panayotis N, Karpova A, Kreutz MR, Fainzilber M. Macromolecular transport in synapse to nucleus communication. Trends Neurosci. 2015;38:108-16
18. Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M. c-fos regulates neuronal excitability and survival. Nat Genet. 2002;30:416-20
19. Westerink RH, Beekwilder JP, Wadman WJ. Differential alterations of synaptic plasticity in dentate gyrus and CA1 hippocampal area of Calbindin-D28K knockout mice. Brain Res. 2012;1450:1-10
20. Kunert S, Meyer I, Fleischhauer S, Wannack M, Fiedler J, Shivdasani RA. et al. The microtubule modulator RanBP10 plays a critical role in regulation of platelet discoid shape and degranulation. Blood. 2009;114:5532-40
21. Cheng PH, Chang YF, Mao SH, Lin HL, Chen CM, Yang SH. Lentiviral transgenesis in mice via a simple method of viral concentration. Theriogenology. 2016;86:1427-35
22. Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594-6
23. Go H, La P, Namba F, Ito M, Yang G, Brydun A. et al. MiR-196a regulates heme oxygenase-1 by silencing Bach1 in the neonatal mouse lung. Am J Physiol Lung Cell Mol Physiol. 2016;311:L400-11
24. Tripurani SK, Lee KB, Wee G, Smith GW, Yao J. MicroRNA-196a regulates bovine newborn ovary homeobox gene (NOBOX) expression during early embryogenesis. BMC Dev Biol. 2011;11:25
25. Mori M, Nakagami H, Rodriguez-Araujo G, Nimura K, Kaneda Y. Essential role for miR-196a in brown adipogenesis of white fat progenitor cells. PLoS Biol. 2012;10:e1001314
26. Yuan Y, Anbalagan D, Lee LH, Samy RP, Shanmugam MK, Kumar AP. et al. ANXA1 inhibits miRNA-196a in a negative feedback loop through NF-kB and c-Myc to reduce breast cancer proliferation. Oncotarget. 2016;7:27007-20
27. Li Y, Jin L, Chen D, Liu J, Su Z, Yang S. et al. Tumor suppressive miR-196a is associated with cellular migration, proliferation and apoptosis in renal cell carcinoma. Mol Med Rep. 2016;14:560-6
28. Tsai MM, Wang CS, Tsai CY, Huang CG, Lee KF, Huang HW. et al. Circulating microRNA-196a/b are novel biomarkers associated with metastatic gastric cancer. Eur J Cancer. 2016;64:137-48
29. Miyazaki Y, Adachi H, Katsuno M, Minamiyama M, Jiang YM, Huang Z. et al. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat Med. 2012;18:1136-41
30. Schulze H, Dose M, Korpal M, Meyer I, Italiano JE Jr, Shivdasani RA. RanBP10 is a cytoplasmic guanine nucleotide exchange factor that modulates noncentrosomal microtubules. J Biol Chem. 2008;283:14109-19
31. Sato Y, Kato A, Maruzuru Y, Oyama M, Kozuka-Hata H, Arii J. et al. Cellular Transcriptional Coactivator RanBP10 and Herpes Simplex Virus 1 ICP0 Interact and Synergistically Promote Viral Gene Expression and Replication. J Virol. 2016;90:3173-86
32. Harada N, Yokoyama T, Yamaji R, Nakano Y, Inui H. RanBP10 acts as a novel coactivator for the androgen receptor. Biochem Biophys Res Commun. 2008;368:121-5
33. Zhao T, Hong Y, Li S, Li XJ. Compartment-Dependent Degradation of Mutant Huntingtin Accounts for Its Preferential Accumulation in Neuronal Processes. J Neurosci. 2016;36:8317-28
34. Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292-309
35. Lin YF, Xu X, Cape A, Li S, Li XJ. Huntingtin-associated protein-1 deficiency in orexin-producing neurons impairs neuronal process extension and leads to abnormal behavior in mice. J Biol Chem. 2010;285:15941-9
36. Kunkanjanawan T, Carter RL, Prucha MS, Yang J, Parnpai R, Chan AW. miR-196a Ameliorates Cytotoxicity and Cellular Phenotype in Transgenic Huntington's Disease Monkey Neural Cells. PloS one. 2016;11:e0162788
Author contact
![]() Corresponding author: Shang-Hsun Yang, Ph.D., Department of Physiology, College of Medicine, National Cheng Kung University, Tainan 70101, Taiwan Email: syangncku.edu.tw Tel:+886-6-2353535 ext.5453 Fax:+886-6-2362780
Corresponding author: Shang-Hsun Yang, Ph.D., Department of Physiology, College of Medicine, National Cheng Kung University, Tainan 70101, Taiwan Email: syangncku.edu.tw Tel:+886-6-2353535 ext.5453 Fax:+886-6-2362780