Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(12):3138-3154. doi:10.7150/thno.19506 This issue Cite
Research Paper
RBFOX3 Promotes Tumor Growth and Progression via hTERT Signaling and Predicts a Poor Prognosis in Hepatocellular Carcinoma
Tianze Liu1, 4*, Wenbin Li1*, Wenjing Lu4*, Miao Chen1*, Meihua Luo2, Changlin Zhang1, Yixin Li1, Ge Qin1, Dingbo Shi1, Binyi Xiao1, Huijuan Qiu1, Wendan Yu3, Lan Kang3, Tiebang Kang1, Wenlin Huang1, 5, Xinfa Yu2 ![]() , Xiaojun Wu1
, Xiaojun Wu1 ![]() , Wuguo Deng1
, Wuguo Deng1 ![]()
1. Sun Yat-sen University Cancer Center; State Key Laboratory of Oncology in South China; Collaborative Innovation Center of Cancer Medicine, Guangzhou, China;
2. ShunDe Hospital of Southern Medical University, Foshan, Guangdong, China;
3. Institute of Cancer Stem Cell, Dalian Medical University, Dalian, China;
4. The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, China;
5. State Key Laboratory of Targeted Drug for Tumors of Guangdong Province, Guangzhou Double Bioproduct Inc., Guangzhou, China.
* These authors contributed equally to this article.
Received 2017-2-5; Accepted 2017-5-19; Published 2017-7-22
Abstract

Activation of the telomere maintenance mechanism is a key hallmark of cancer. Human telomerase reverse transcriptase (hTERT) is the catalytic subunit of telomerase, which is highly expressed in more than 80% of tumors, including hepatocellular carcinoma (HCC). However, the exact mechanisms by which hTERT is up-regulated in HCCs and promotes tumor growth and progression is not fully understood. The aim of this study was to discover the novel molecular targets that modulate hTERT signaling and HCC growth. In this study, we pulled down and identified RBFOX3 (RNA binding protein fox-1 homolog 3) as a novel hTERT promoter-binding protein in HCC cells using biotin-streptavidin-agarose pull-down and proteomics approach, and validated it as a regulatory factor for hTERT signaling and tumor growth in HCCs. Knockdown of RBFOX3 suppressed the promoter activity and expression of hTERT and consequently inhibited the growth and progression of HCC cells in vitro and in vivo. The suppression of HCC growth mediated by RBFOX3 knockdown could be rescued by hTERT overexpression. Conversely, exogenous overexpression of RBFOX3 activated the promoter activity and expression of hTERT and promoted the growth and progression of HCC cells. Moreover, we found that RBFOX3 interacted with AP-2β to regulate the expression of hTERT. Furthermore, we demonstrated that RBFOX3 expression was higher in the tumor tissues of HCC patients compared to the corresponding paracancer tissues, and was positively correlated with hTERT expression. Kaplan-Meier analysis showed that the HCC patients with high levels of RBFOX3 and hTERT had poor prognosis. Collectively, our data indicate that RBFOX3 promotes HCC growth and progression and predicts a poor prognosis by activating the hTERT signaling, and suggest that the RBFOX3/hTERT pathway may be a potential therapeutic target for HCC patients.
Keywords: HCC, telomerase reverse transcriptase, promoter-binding protein.
Introduction
Hepatocellular carcinoma (HCC), the fifth most common cancer, is extremely aggressive and associated with poor prognosis, and remains the third most common cause of cancer-related deaths all over the world [1, 2]. Moreover, the incidence of HCC has been rising steadily in the recent 20 years predominantly due to increased rates of chronic Hepatitis C virus infections and alcohol abuse [3]. Despite recent advances in therapeutic strategies, the long-term prognosis of patients with HCC remains poor. Therefore, it is urgent to further understand the molecular mechanisms in HCC tumorigenesis and progression and to discover and identify new therapeutic targets for this tumor.
Telomerase, a repetitive nucleoprotein structure, adds the 5'-TTAGGG-3' repeats onto the ends of linear chromosomes and provides a telomere maintenance mechanism for about 85-90% of human cancers and eukaryotic cells [4, 5]. In the absence of telomerase, telomeres undergo progressive shortening during successive replication cycles, and eventually reach critically short lengths that result in cellular DNA damage or apoptosis [6]. Accordingly, telomerase has been found to play important roles in tumorigenesis and development. Telomerase is a ribonucleoprotein complex consisting of RNA and human telomerase reverse transcriptase (hTERT). As the catalytic subunit of telomerase, hTERT contains conserved catalytic reverse transcriptase motifs and determines the activity of telomerase [7-10]. When telomerase is activated in the cell, hTERT synthesizes a TTAGGG sequence from the RNA template, which is then added to the ends of the shortening telomeres, and saves the cells from aging or death [11]. Therefore, hTERT is a potential target for telomerase inhibition. Overexpression of hTERT is a common feature of nearly all human cancers and is believed to support cell immortalization [12], as the expression of hTERT is usually low or hardly detected in normal human cells except embryonic stem cells and germ cells. However, it is still unclear why hTERT expression is low in normal cells but elevated during the process of carcinogenesis. Multiple transcription factors have been reported to bind to the hTERT promoter region to control its expression [13], and we speculate that certain transcription factors or regulatory factors differentially or selectively bind to the hTERT promoter region in human cancer cells to promote hTERT expression, thus regulating tumor development.
RNA binding protein fox-1 homolog 3 (RBFOX3) is encoded by the Rbfox3 gene that is located on chromosome 17 and comprises 15 exons, and is a novel member of the fox1 family of splicing factors [14]. The fox1 family has three members in mammals, RBFOX1, RBFOX2, and RBFOX3. RBFOX1 is expressed in heart, skeletal muscles, and neural tissues. RBFOX2 has a broad expression profile in different tissues, including throughout the embryo, neurons, and muscles. However, RBFOX3 is primarily expressed in post-mitotic neurons in normal tissues [15, 16]. It is well established that RBFOX3 regulates a battery of brain-specific alternative pre-mRNA splicing choices by binding to an RNA penta(hexa)nucleotide (U)GCAUG motif [17], and a study has revealed that RBFOX3 interacts with the polypyrimidine tract binding protein-associated splicing factor (PSF) [18], which enhances the binding of RBFOX3 to the target UGCAUG element [19]. However, recent studies have indicated that RBFOX3 is not just an alternative splicing factor, but is also involved in physiological processes and certain disease events. Kim's work showed that neuronal nuclei (NeuN) was also the product of the Rbfox3 gene [20], revealing that RBFOX3 was a well-recognized “marker” that is detected exclusively in post-mitotic neurons. Another study showed that RBFOX3 could bind to DNA in vitro [21]. Kim et al. found RBFOX3 could control the biogenesis of a subset of microRNAs including primary-microRNAs (pri-miRNAs) that lack the UGCAUG motif [22]. Based on the research progresses of RBFOX3 above, we have reasons to believe that RBFOX3 does not only function in alternative splicing of pre-mRNAs to regulate gene expression post-transcriptionally, but also plays critical roles in other biochemical aspects that are still unclear. Here, we have discovered and identified that RBFOX3 has a new molecular feature in binding at the promoter of hTERT to modulate hTERT expression and regulate cell growth.
In this study, we used biotin-streptavidin-agarose pull-down assay, an approach for analyzing the binding of an array of proteins on a DNA sequence [23, 24], to find proteins bound at the promoter region of hTERT in hepatocellular carcinoma cells. We identified RBFOX3 as a novel hTERT promoter-binding protein, and further demonstrated that RBFOX3 bound to the endogenous hTERT promoter in HCC cell lines by chromatin immunoprecipitation assay. Our results showed that, the binding of RBFOX3 at the hTERT promoter activated hTERT expression in HCC cells, thereby promoting HCC cell growth and progression. Furthermore, we found RBFOX3 interacted with AP-2β to regulate the expression of hTERT. Our results were confirmed by an in vivo tumor model, and the expression status and clinical significance of RBFOX3 in HCC were also investigated. Our study therefore demonstrated that RBFOX3 regulated HCC carcinogenesis and development indirectly through the activation of hTERT, and suggested that the RBFOX3/hTERT signaling pathway could serve as a potential novel therapeutic target for hepatocellular carcinoma.
Materials and Methods
Cell lines and antibodies
Human hepatocellular carcinoma cells (Hep3B, QGY7703, HepG2, and SNU-449), N9 microglia cell (N9 MG) cell and glioma cell lines (U138, U251 and U373) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco's Modified Eagle Medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum. Human immortalized hepatic epithelial cell line LO2 was cultured in RPMI1640 (Gibco BRL, Grand Island, NY) with 10% fetal bovine serum. All cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. RBFOX3 antibodies for Western blot, ChIP and immunofluorescence staining were purchased from Sigma (sab4301175), Merck Millipore (MAB377), and Cell Signaling Technology (12943s), respectively. Other antibodies were purchased from Cell Signaling Technology.
Streptavidin-agarose pulldown assay
The hTERT promoter binding proteins were analyzed by streptavidin-agarose pulldown assay as described previously [23]. Briefly, 1 mg of nuclear protein extracts from human hepatocellular carcinoma cells were incubated with 10 µg of biotin-labeled double-stranded DNA probes corresponding to nucleotide -378 to -157 of the hTERT promoter region (Sigma-Aldrich, St Louis, MO) and 100µl of streptavidin-agarose beads (Sigma-Aldrich) at 4°C overnight. The mixture was then centrifuged at 500 × g to pulldown the DNA-protein complex.
Identification of hTERT promoter-binding proteins
Proteins bound on the hTERT promoter pulled down by streptavidin-agarose beads were analyzed by mass spectrometry. Briefly, the bound proteins were separated by 10% SDS-PAGE and visualized by sliver staining (Beyotime, Shanghai, China). After reduction and alkylation, the candidate protein bands were digested with MS-grade trypsin solution (Promega, Madison, WI), and the digested peptides were identified by mass spectrometry. The identities of the proteins of interest were verified via available databases and software.
Transient transfection
To overexpress RBFOX3 and AP-2β in HCC cells, pcDNA3.1-RBFOX3, pcDNA3.1-AP-2β or control vector plasmids were transfected with Lipofectamine 3000 (Invitrogen, Carlsbad, CA). To inhibit RBFOX3, AP-2β, RBFOX1, and RBFOX2 expression, HCC cells were transfected with RBFOX3 specific short hairpin RNA (shRNA, 5'-GCG GCA AAT GTT CGG GCA A-3' and 5'-GGC TAC ACG TCT CCA ACA T-3'), RBFOX1 specific siRNA (5'-GCA CGU GUA AUG ACA AAU ATT-3' and 5'-GAG CCU GUG UAU GGC AAU ATT-3'), RBFOX2 specific siRNA (5'-GCC ACA CAC UCA AGA CUA UTT-3' and 5'-GCU GCA UGU CUC UAA UAU UTT-3'), and AP-2β specific siRNA (5'-GGA CCA GUC UGU CAU UAA ATT-3'), respectively. siRNAs were purchased from Shanghai GenePharma Co. (Shanghai, China).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed according to Carey's protocol. Briefly, the cells were fixed with 1% formaldehyde, and the cross-linking was quenched by adding in 100 μl of 1.375 M glycine per milliliter of culture. The samples were sonicated on ice to shear the DNA into 300 to 1000 bp fragments. For each total cell lysate, one third was used as the DNA input control, another third was immunoprecipitated with anti-RBFOX3 antibodies, and the last third was subjected to non-immune rabbit IgG (Cell Signaling Technology, Danvers, MA). DNA fragments were purified by spin columns (Qiagen, Hilden, Germany), and PCR was performed to amplify a 230-bp segment in the promoter region of hTERT with the following primer pair -Forward: 5'-TGGCCCCTCCCTCGGGTTAC-3', Reverse: 5'-TGAAG GGGCAGGACGGGTGC-3'. The PCR products were resolved by electrophoresis in a 2% agarose gel and visualized by Gel-Red staining.
Lentiviral Construction and Cell Transfection
To generate clones stably overexpressing RBFOX3, SNU-449 and QGY-7703 cells were infected with a lentiviral vector encoding a full-length human RBFOX3 gene or an empty lentiviral vector control. Stable clones were selected after 2 weeks using 0.7-5 µg/ml puromycin and the expression level of RBFOX3 was determined by qRT-PCR and Western blot.
The U6-sh-RBFOX3-EGFP-IRES-puromycin vectors were obtained from Genechem Company Ltd (Shanghai, China) and used to knock down RBFOX3 expression. A lentiviral vector containing non-silencing short hairpin RNA was used as the negative control. HepG2 and Hep3B cells were infected with either the lentiviral vectors encoding specific shRNA sequences or the negative control vector. The efficiencies of RNA interference were determined by qRT-PCR and Western blot.
Promoter reporters and dual-luciferase assay
A fragment containing the core promoter region of hTERT (-387-+40) was inserted between the SacI and HindІІІ sites of the firefly luciferase vector pGL4.10 (Promega, Madison, WI), and Renilla luciferase control reporter vector pRL-TK was used as a control. Cells (2×104 cells/well) were seeded in a 96 wells plate. After the cell confluence reached 50%, pGL4.10-hTERT-387 or pRL-TK was transfected into HepG2 and Hep3B cells with RBFOX3 stable knockdown or overexpression with Lipofectamine 3000 (Invitrogen, Carlsbad, CA) (30:1 ratio). 36 hours after the transfection, dual-luciferase assay was performed using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI).
Cell viability assay
HCC cells were seeded at a density of 3×103 cells per well in 96-well plates, and the viability of the cells was assessed by the MTS assay (Promega, Madison, WI) 72 hours later. The absorbance value at 490 nm in each well was measured with a microplate reader. All experiments were performed in 6 replicates per trial, with 3 independent trials in total and the average percentages of cell viability were shown.
Colony formation assay
HCC cells were seeded at a density of 200-400 cells per well in 6-well plates and cultured for 2 weeks. The colonies were then stained with 1% crystal violet and counted. All experiments were performed with 3 independent trials.
Wound healing and trans-well invasion assays
To determine the mobility and invasion of HCC cells in the conditions of BRFOX3 knockdown and overexpression, wound healing assay and trans-well invasion assay were performed respectively. Briefly, HCC cells with stable overexpression or knockdown of BRFOX3 or the control vector were cultured in 6-well plates till confluence, and then scratched with a 10 μl pipette tip. Cell migration images were captured at 0h and 24h after scratching. Each sample was analyzed in triplicates. Cell invasion assay was performed with BD BioCoat Matrigel Invasion Chambers (Becton Dickinson, Franklin Lakes, NJ) per manufacturer's instruction. Five random fields were counted under the light microscope.
Western blot
HCC cells were infected with lentivirus and screened by puromycin after two weeks. Whole cell lysates or nuclear extracts were prepared using Complete Lysis-M reagent (Roche, USA) and RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China). Protein concentration was determined by BCA assay (ThermoFisher Scientific, Waltham, MA). The proteins were separated in 8%-10% SDS-PAGE gels and transferred onto PVDF membranes for detection. Antibodies for RBFOX3, Histone3, GAPDH, PI3K, p-PI3K, AKT, p-AKT, JNK, p-JNK, ERK, p-REK, MMP2, MMP9, Cyto-C, caspase 7, PARP, caspase 9 and caspase 3 were purchased from Cell Signaling Technology (Danvers, MA). Anti-TERT rabbit antibody is purchased from Abcam (Cambridge, UK).
Real time PCR (qPCR)
Total RNA was isolated using TRIZOL Reagent (Invitrogen, Carlsbad, CA) per instruction. cDNA was synthesized using the ReverTra Ace qPCR RT Master Mix (Toyobo, Japan). The SYBR Green PCR master mix (Toyobo, Osaka, Japan) was then used for qPCR, which was followed by detection with a Bio-Rad CFX96 and analyzed with the Bio-Rad Manager software (Bio-Rad, Hercules, CA). Expression level relative to RBFOX3 or hTERT was calculated by 2-ΔCT (ΔCT = CTRBFOX3 or hTERT - CTGAPDH) and normalized to the relative expression level detected in control cells. Each sample was tested in triplicate. Primers were purchased from GeneCopoeia (TERT, Hs-QRP22639; GAPDH, Hs-QRP20169; RBFOX1, Hs-QRP22117; RBFOX2, Hs-QRP49512; RBFOX3, Hs-QRP22696).
Apoptosis and cell cycle analysis
At 72 h after transfection, cells (1×105 cells/ml) were stained with 5 ml AnnexinV-FITC and 5 ml PI (propidium iodide), incubated in room temperature for 15 min in the dark, and then analyzed by flow cytometry (EPICS XL, Beckman Coulter, Brea, CA). Apoptosis was determined as FITC-positive in cells. Cell cycle analysis was performed using PI staining.
Co-immunoprecipitation assays
Equal amounts of nuclei protein extracts prepared from different cell lines were incubated with the indicated antibodies. Then, the agarose-conjugated protein-A/G beads (Merck Millipore, Billerica, MA) were added and the mixture was incubated at 4°C overnight. After extensive washing with cold phosphate-buffered saline (PBS), the beads were mixed with loading buffer and boiled. The proteins in the supernatant were detected by Western blotting analysis.
Flag-tagged RBFOX3 protein purification
The cells overexpressing flag-tagged RBFOX3 were lysed with cell lysis buffer containing 0.1% tween 20 and 0.1% triton-100 to break the interaction between proteins. The anti-flag G1 affinity resin was added for binding flag-tagged RBFOX3 protein. After washing, flag-tagged proteins were eluted with the DYKDDDDK peptide. The purified protein was separated by SDS-PAGE and analyzed by silver staining.
In vivo tumor model and tissue processing
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publications Nos. 80-23, revised 1996) and the Institutional Ethical Guidelines for Animal Experiments developed by Sun Yat-sen University. Six groups of 5 female nude mice (4-5 weeks old and 15-23 g) were subcutaneously injected with 3×106 HCC cells. The length (L), width (W) and height (H) of each tumor was measured with calipers, and the volume (V) was calculated as follows: V=πLWH/6.
The RBFOX3 and hTERT expression in HCC patients' tumor tissues and mouse xenografts tumor tissues were measured using IHC staining. Briefly, after deparaffinizing, blocking and antigen retrieval, the tumor sections were then incubated in 1:50 dilution of RBFOX3-specific antibody (Sigma) or 1:40 dilution of TERT-specific antibody (Nouvs Biologicals, Littleton, CO) at 4°C overnight in a humidified chamber. After washing, tumor sections were incubated with horseradish peroxidase-conjugated anti-goat antibody for 30 min at room temperature and were developed color with 3,5-diaminobenzidine (DAB) substrate followed by Mayer's hematoxylin counterstaining. For IHC score, the percentage (0-100%) of stained tumor cells was multiplied by the intensity (0, 1, 2, or 3) to achieve a score between 0 and 300.
Metastasis assay
Four groups of 6 mice were each given intravenous tail vein injection with 2×106 HCC cells. After 8 weeks of observation, mice were sacrificed and tumor nodules on the lung surfaces were counted, excised and embedded in paraffin.
Statistical analysis
Data were presented as mean ± standard deviation from at least three independent experiments. Statistical analysis was carried out using SPSS 11.0 software (SPSS Inc.; Chicago, IL). P< 0.05 was considered statistically significant.
Results
RBFOX3 Was Identified as an hTERT Promoter-Binding Protein in HCC cells
In our previous study, we identified novel regulators of the hTERT promoter using streptavidin-agarose bead pulldown method in lung cancer [23]. In this study, to screen and identify differential hTERT promoter binding proteins in HCC cells, a 5'-biotin labeled 230-bp DNA probe for the region of -387 to -157 of the hTERT promoter was synthesized (Supplementary Figure 1A). We incubated this probe with nuclear protein extracts from four human HCC cell lines (Hep3B, QGY7703, HepG2, SNU-449) and one immortalized liver cell line (LO2) to pull down hTERT promoter binding proteins, which were then separated by SDS-PAGE. Silver staining of the protein gel showed that one of the protein bands (at approximately 34 kDa) was significantly elevated in HCC cells in comparison with immortalized liver cells (Figure 1A, arrow).
RBFOX3 was identified as an hTERT promoter binding protein in HCC cells. (A) The potential hTERT promoter binding proteins were pulled down using 5'-biotin labeled hTERT promoter DNA probe (-157 to -387) in four HCC cell lines (Hep3B, QGY7703, HepG2 and SNU-449) and immortalized liver cells (LO2). The proteins were separated by SDS-PAGE, and visualized using sliver staining. Arrow showed the target protein band was significantly enriched in Hep3B, QGY7703, HepG2 and SNU-449 cells in comparison with LO2. (B) The protein band indicated with the red arrow in Figure 1A was excised, trypsinized and analyzed by MALDI-TOF/TOF mass spectrometry. The identified peptides were shown in Figure 1B, and the assigned b and y ion peaks on the spectrum were marked with their corresponding m/z values. (C) Binding of RBFOX3 on the 5'-biotin labeled hTERT promoter probe or a nonspecific probe (NSP) was detected by Western blot using anti-RBFOX3 antibody. RBFOX3 protein was detected in the nuclear protein-hTERT probe-streptavidin bead complexes in Hep3B, QGY7703, HepG2 and SNU-449 cells, but very little RBFOX3 was detected in LO2 cells (upper panel). The expression of total RBFOX3 proteins in HCC cells were analyzed by Western blot, and GAPDH was used as a loading control (lower panel). (D) Chromatin immunoprecipitation (ChIP) assays were performed in HCC and immortalized liver cells using RBFOX3 antibody and the hTERT promoter primers. The PCR products were separated in 2% agarose gels. IgG was used as a negative control. (E) ChIP assays were carried out using the hTERT promoter primers and RBFOX3 antibody in QGY7703 cells with RBFOX3 overexpression or vector plasmid (lower panel) and Hep3B cells with sh-RBFOX3 or sh-NC (upper panel). M, Mock, transfection reagents control; shNC, non-sense shRNA control; RBFOX3, RBFOX3 overexpression. (F) The 5'-biotin labeled probes corresponding different fragment of hTERT promoter (probe 1-7, left panel) or a nonspecific probe (NSP) were incubated with Hep3B cell lysates and streptavidin beads. The bound proteins were detected by Western blot using anti-RBFOX3 antibody (right panel). (G) The protein RBOFX3 was purified with anti-flag antibody affinity resin with lysates from Hep3B/HepG2-control and Hep3B/HepG2-Flag- RBFOX3 cells. The bound proteins were eluted with flag peptide and analyzed by silver staining (left panel). The purified protein was pulled down using 5'-biotin labeled hTERT promoter probe or a nonspecific probe (NSP) and streptavidin beads, and the bound proteins was detected by Western blot using anti-RBFOX3 antibody (right panel).
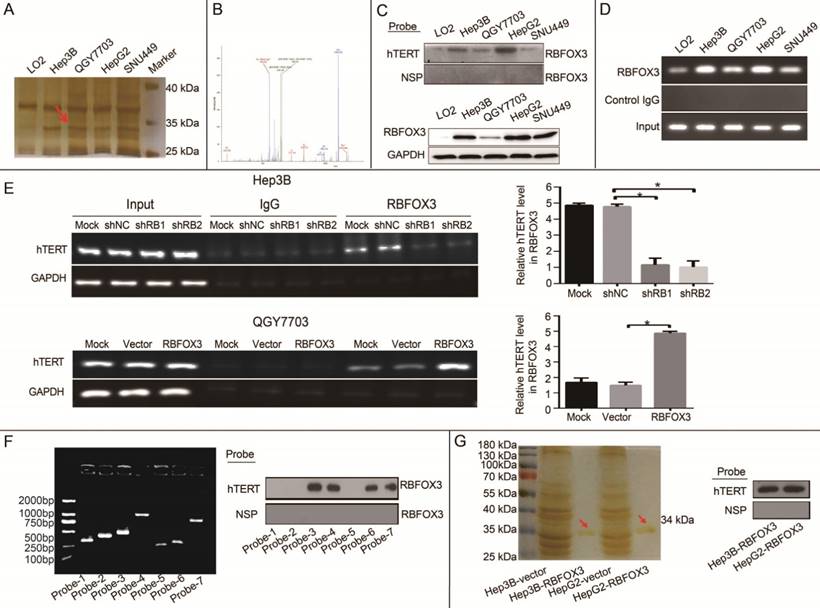
To identify the special candidate hTERT promoter binding protein in HCC cells, the protein band indicated with the red arrow in Figure 1A was excised, trypsinized and analyzed by MALDI-TOF/TOF mass spectrometry. The identified peptides were shown in Figure 1B, and the assigned b and y ion peaks on the spectrum were marked with their corresponding m/z values, which were compared with the theoretical m/z values of the amino acid residues in Supplementary Table 1 and identified to be VNNATARVMTNK. We then searched for this peptide sequence in the proteomics database (https://blast.ncbi.ncbi.ncm.nih.gov/Blast.cgi? PROGRAM=blastp&PAGE_TYPE=BlastSearch&LIK_LOC=blasthome), and found it to align with the sequence of RNA binding protein fox-1 homolog 3 (RBFOX3).
To further validate the interaction between RBFOX3 and the hTERT promoter, we pulled down the nuclear proteins bound at the hTERT promoter in HCC cells using the 5'-biotin labeled hTERT promoter probe and streptavidin-agarose beads, and detected RBFOX3 in the nuclear protein/DNA complex using the RBFOX3-specific antibody by Western blot. High level of RBFOX3 was bound to the hTERT promoter probe in HCC cells (Hep3B, QGY7703, HepG2, and SNU-449). However, very little RBFOX3 was bound at the hTERT promoter probe in LO2 (Figure 1C, upper panel). We also investigated the level of RBFOX3 bound to the hTERT promoter probe in the condition of RBFOX3 overexpression or knockdown. RBFOX3 bound to the hTERT promoter probe was increased in RBFOX3 overexpressing QGY7703 cells, but decreased in RBFOX3 knockdown Hep3B cells (Supplementary Figure 1B and 1C). The expression of total RBFOX3 in cell lines showed Hep3B and HepG2 cells had higher RBFOX3 expression than QGY7703 and SNU-449 cells, while the expression of RBFOX3 proteins in LO2 was very low (Figure 1C, lower panel). Furthermore, to confirm that RBFOX3 interacted with the hTERT promoter in vivo, ChIP assay was performed in HCC cells and immortalized liver cells. We found the RBFOX3 protein bound to the endogenous hTERT promoter in all cell lines in the nucleus. Importantly, very weak binding of RBFOX3 to the hTERT promoter was observed in LO2 cells, but strong binding of RBFOX3 to the hTERT promoter was detected in all four human HCC cells (Hep3B, QGY7703, HepG2, and SNU-449) (Figure 1D). Consistently, RBFOX3 bound weakly to the hTERT promoter in Hep3B cells with RBFOX3 knockdown, but bound strongly to the hTERT promoter in RBFOX3 overexpressing QGY7703 cells (Figure 1E).
To identify the binding region of RBFOX3 in the hTERT promoter, seven different fragments of 5'-biotin labeled hTERT promoter (Supplementary Figure 1D and Figure 1F) were constructed and incubated with Hep3B cell lysates. The pulldown result indicated that the -387 to -321 (5'-CTC GGG TTA CCC CAC AGC CTA GGC CGA TTC GAC CCT CTC CGC TGG GGC CCT CGC TGG CGT CCC TGC-3') region of hTERT promoter was critical for RBFOX3 binding. To further study whether RBFOX3 bound to the hTERT promoter directly, protein extracts prepared from flag-RBFOX3 overexpressing Hep3B and HepG2 cells were purified with GenScript Anti-DYKDDDDK G1 Affinity Resin (Cat. No. L00432). The result of silver staining showed the purified RBFOX3 protein at around 34 kDa (Figure 1G, left panel). The purified protein was incubated with hTERT promoter probe, and the pulldown result showed that the purified RBFOX3 could still bind to the hTERT promoter, which indicated that RBFOX3 bound to the hTERT promoter directly (Figure 1G, right panel).
RBFOX3 Enhanced hTERT Promoter Activity and Expression in HCC Cells
Next, we investigated the effect of RBFOX3 on hTERT promoter activity and gene expression. We constructed the RBFOX3 overexpressing plasmids and synthesized two RBFOX3 short hairpin RNAs (shRNAs). The luciferase reporter assay showed that overexpression of RBFOX3 dramatically enhanced the hTERT promoter activity in contrast to the vector group in QGY7703 and SNU-449 cells, whereas knockdown of RBFOX3 significantly decreased the hTERT promoter activity in HepG2 and Hep3B cells in contrast to the non-specific shRNA control (shNC) groups (Figure 2A and Supplementary Figure 2A). We also found that overexpression of RBFOX3 promoted hTERT mRNA and protein levels in QGY7703 and SNU-449 cells, while knockdown of RBFOX3 inhibited hTERT expression at transcriptional and translational levels in HepG2 cells and Hep3B cells (Figure 2B and2C, Supplementary Figure 2B and2C).
RBFOX3 regulated hTERT promoter activity and expression in HCC cells. (A) Relative hTERT promoter activity in RBFOX3 overexpressing QGY7703 cells and RBFOX3 knockdown Hep3B cells. (B) Relative hTERT mRNA level was determined by quantitative real-time PCR analyses in RBFOX3 overexpressing QGY7703 cells and RBFOX3 knockdown Hep3B cells. (C) hTERT protein expression was up-regulated in RBFOX3 overexpressing QGY7703 cells, and down-regulated in RBFOX3 knockdown Hep3B cells. (D) The expression of total RBFOX3 proteins in N9 microglia (N9 MG) cell line, glioma cell lines (U138, U251 and U373), and HCC cell lines (Hep3B, QGY7703 and HepG2) were analyzed by Western blot. GAPDH was used as a loading control. (E) hTERT protein expression was detected with RBFOX3 knockdown in N9 MG and U251 cell lines. (F) ChIP assays were performed in neuronal cells and HCC cells using RBFOX3 antibody and the hTERT promoter primers. The PCR products were separated in 2% agarose gels. The IgG was used as a negative control. (G) Relative hTERT mRNA and protein levels were determined in RBFOX1 knockdown Hep3B cell lines by quantitative real-time PCR and Western blot, respectively. (H) Relative hTERT mRNA and protein levels were determined in RBFOX2 knockdown Hep3B cell lines by quantitative real-time PCR and Western blot, respectively. Results were shown as means ± SD, *p<0.05 by two-tailed Student's t-test. All experiments were performed in at least 3 independent trials.
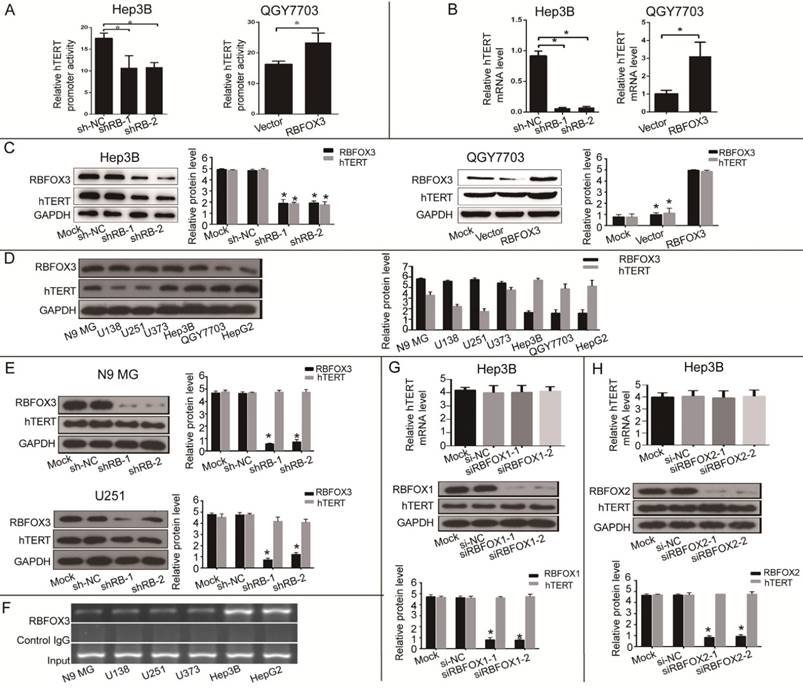
As RBFOX3 is a well-recognized neuronal marker, we wondered whether RBFOX3-mediated hTERT gene regulation also occurred in the neuronal cells. We detected the expression of RBFOX3 and hTERT in N9 microglia (N9 MG) and glioma cell lines (U138, U251 and U373). As was shown in Figure 2D, compared with HCC cells, neuronal cells had higher expression of RBFOX3 but lower expression of hTERT. And we found that knockdown of RBFOX3 slightly down-regulated the expression of hTERT in U251 cell line. However, the change of hTERT expression was not obvious when the RBFOX3 was knockdown in N9 MG, U373, and U138 cell lines (Figure 2E and Supplementary Figure 2D). Furthermore, ChIP assay showed the binding of RBFOX3 to the hTERT promoter in vivo was much weaker in neuronal tumor cell lines than in HCC cells (Figure 2F). Therefore, we proposed that RBFOX3 might not be the major transcription regulator of hTERT in the neuronal tumor cell lines.
In addition, we also investigated whether both RBFOX1 and RBFOX2, the RBFOX3 family proteins, could regulate the transcription and translation of hTERT. The results showed that neither RBFOX1 (Figure 2G and Supplementary Figure 2E) nor RBFOX2 (Figure 2H and Supplementary Figure 2F) could regulate the expression of hTERT at transcriptional and translational levels in HCC cells. These results suggested that RBFOX3 specifically activated hTERT expression in HCC cells.
RBFOX3 Promoted HCC Growth in vitro and in vivo via hTERT Signaling Pathway
To further investigate the function of RBFOX3 in HCC cell growth in vitro and in vivo, we established the RBFOX3 overexpression stable cell lines (SNU-449, QGY7703) and RBFOX3 knockdown stable cell lines (HepG2, Hep3B). We found that overexpression of RBFOX3 significantly enhanced HCC cell viability and colony formation in QGY7703 cells (Figure 3A) and SNU-449 cells (Supplementary Figure 3A). We also found that knockdown of RBFOX3 effectively inhibited HCC cell viability and colony formation in Hep3B cells (Figure 3B) and HepG2 cells (Supplementary Figure 3B). Moreover, this inhibition mediated by knockdown of RBFOX3 was reversed by hTERT overexpression in Hep3B (Figure 3C) and QGY7703 cells (Supplementary Figure 3C). Then we performed the hTERT expression rescue experiments for RBFOX3 overexpression in RBFOX3-depleted Hep3B and QGY7703 cells. We found that the expression of hTERT was down-regulated in RBFOX3-depleted cells, while overexpression of RBFOX3 rescued the expression of hTERT (Figure 3D and Supplementary Figure 3D). These results indicated that both endogenous and exogenous expression of RBFOX3 could regulate the expression of hTERT. We further checked the effect of RBFOX3 on the pivotal proteins in the PI3K/ERK signaling, a crucial downstream pathway associated with the hTERT signaling, and found that overexpression of RBFOX3 up-regulated p-PI3K, p-AKT, p-JNK and p-ERK expression, but not total PI3K, AKT, JNK and ERK in QGY7703 (Figure 3E) and SNU-449 (Supplementary Figure 3E) cells. These results indicated that RBFOX3 could promote HCC cell growth via hTERT signaling pathway in vitro.
RBFOX3 regulated HCC cell growth via hTERT signaling pathway. (A) RBFOX3 overexpression increased propagative cell viability (left panel) and colony formation capacity (middle panel) in QGY7703 cells. Right panel showed the quantification of colonies. (B) RBFOX3 depletion decreased cell viability (left panel) and colony formation capacity (middle panel) in Hep3B cells. Right panel showed the quantification of colonies. (C) Overexpression of hTERT reversed the inhibition of hTERT expression, cell viability and colony formation mediated by RBFOX3 knockdown in Hep3B cells. Cell viability was determined by absorbance at the wavelength of 490 nm and normalized to the absorbance value in the control group by MTS assay. The absorbance value was shown as means ± SD. (D) Overexpression of RBFOX3 reversed the inhibition of hTERT expression by RBFOX3 knockdown in Hep3B cells. (E) RBFOX3 regulated the expression of the PI3K/AKT signaling proteins. Overexpression of RBFOX3 activated hTERT downstream PI3K/AKT signaling pathway in QGY7703 cells. The protein level of p-PI3K, p-AKT, p-JNK, p-ERK and total PI3K, AKT, JNK and ERK were detected and quantified in RBFOX3 overexpressing QGY7703 cells, GAPDH was used as a loading control. (F) QGY7703 cells with RBFOX3 overexpressing plasmid or vector control were subcutaneously implanted into nude mice. The tumor grafts were excised 21 days after inoculation. (G) Hep3B cells with sh-RBFOX3, sh-NC, sh-RBFOX3+hTERT or sh-RBFOX3+vector were subcutaneously implanted into nude mice. The tumor grafts were harvested 21 days after inoculation.
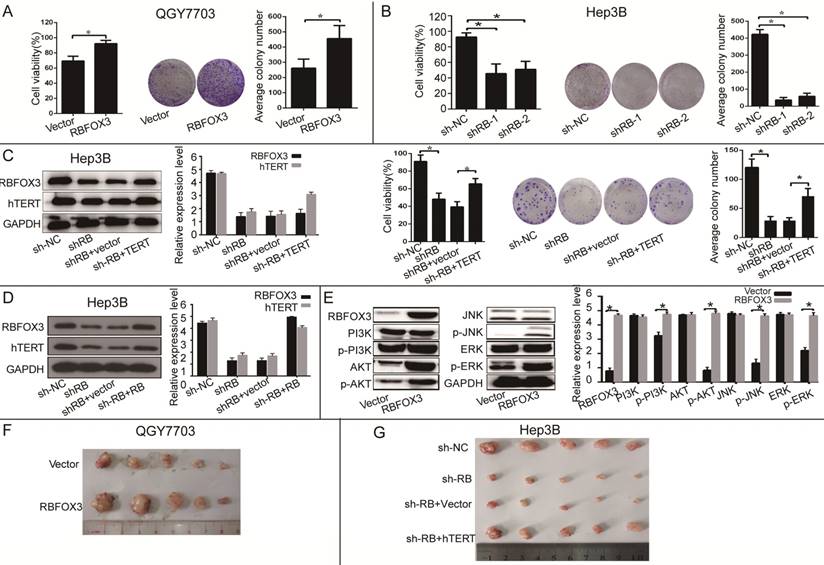
To further demonstrate that RBFOX3 can regulate HCC cell growth in vivo, we established an HCC xenograft model in nude mice. Hep3B cells with stable expression of RBFOX3-shRNAs or the control shRNAs and QGY7703 cells with stable expression of RBFOX3 or the control vector were injected into nude mice. We also transfected hTERT plasmids into RBFOX3 knockdown cells and injected them into nude mice to establish a rescue model. The tumor cell growth was monitored over a period of three weeks. QGY7703 xenografts with RBFOX3 overexpression grew faster than those with the control vector (Figure 3F and Supplementary Figure 3F). In contrast, RBFOX3 knockdown Hep3B xenografts grew at a slower rate in tumor size and weight than the controls, but xenografts with RBFOX3 knockdown and hTERT overexpression had faster growth in tumor size and weight than those with RBFOX3 knockdown and the control vector (Figure 3G and Supplementary Figure 3G). None of these treatments in nude mice affected their body weight or health conditions (data was not shown). Taken together, these results indicated that RBFOX3 promoted HCC cell growth via hTERT in vitro and in vivo.
RBFOX3 Regulated HCC Cell Migration and Invasion via hTERT Signaling Pathway in vitro and in vivo
hTERT signaling pathway is crucial for tumor cell migration and invasion, so we surveyed the function of RBFOX3 in the processes of HCC cell migration and invasion using wound healing and trans-well assays. We found that overexpression of RBFOX3 increased the migration and invasion rate in QGY7703 and SNU-449 cells (Figure 4A and supplementary Figure 4A). We also detected the expression of two members of the MMP family, MMP2 and MMP9, which are vital to cell migration and invasion. We found that overexpression of RBFOX3 in QGY7703 and SNU-449 cells up-regulated MMP2 and MMP9 protein levels (Figure 4C, Supplementary Figure 4C). However, knockdown of RBFOX3 decreased the migration rate in Hep3B and HepG2 cells (Figure 4B and supplementary Figure 4B), and the protein levels of MMP2 and MMP9 were down-regulated simultaneously (Figure 4D and Supplementary 4D). Moreover, overexpression of hTERT in RBFOX3-knocked down cells increased the migration and invasion rates when compared to the RBFOX3-knocked down cells with the control vector (Figure 4E, 4F and Supplementary Figure 4E, 2F).
RBFOX3 regulated HCC cell migration and invasion in vivo and in vitro via hTERT signaling pathway. (A) The relative cell migration and invasion ratio were increased in RBFOX3 overexpressed cells compared with vector overexpressed cells in QGY7703 cells. (B) The relative cell migration and invasion ratio were effectively decreased in RBFOX3 knockdown Hep3B cells. (C) MMP9 and MMP2 protein level were detected and quantified in RBFOX3 or vector overexpressed QGY7703 cells. (D) MMP9 and MMP2 protein level were detected and quantified in sh-RBFOX3 or sh-NC treated Hep3B. (E) Overexpression of hTERT reversed the inhibition of cell migration and invasion by RBFOX3 knockdown in QGY7703 cells. (F) Overexpression of hTERT reversed the inhibition of cell migration and invasion by RBFOX3 knockdown in Hep3B cells. (G) Arrows showed the representative results of metastatic lung nodules, and H&E staining was used to stain metastatic lung nodules (upper panels, scale: 40×; lower panel, scale: 100×). The right panel illustrated the statistical results (n=5). (H) Arrows showed the representative results of metastatic lung nodules, and H&E staining was used to stain metastatic lung nodules (upper panel, scale: 40×; lower panel, scale: 100×). The right panels illustrated the statistical results (n=5). The cell migration capacity was analyzed by scratch assay and the cell invasion capacity was determined by trans-well assay. Data were shown as means ± SD. n = 5 for each group. *p<0.05 by two-tailed Student's t-test.
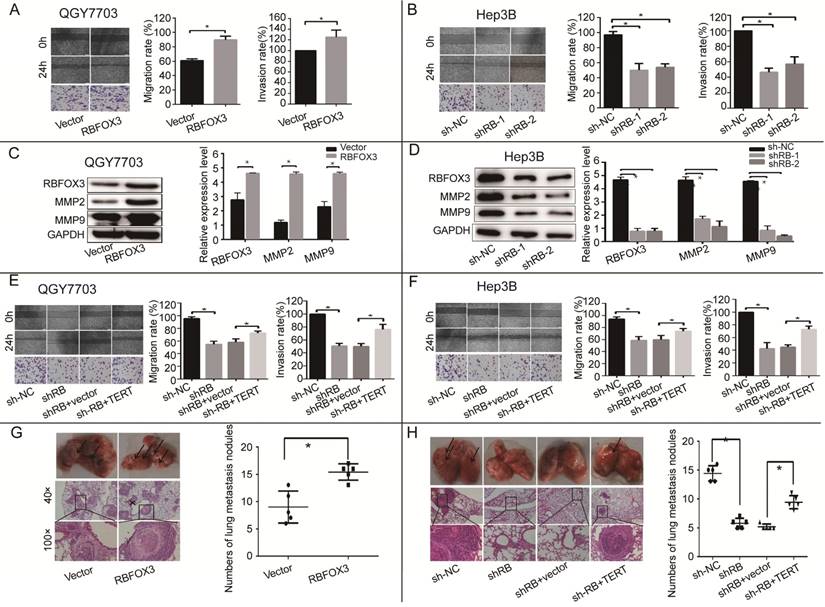
To further confirm that RBFOX3 can regulate HCC cell migration in vivo, a lung metastasis animal model was established through tail vein injection. HE staining was used to confirm pathological metastasis to the lungs, and the number of metastatic tumors was counted to evaluate the effect of RBFOX3. We found that overexpression of RBFOX3 promoted the formation of metastatic tumors in the lungs (Figure 4G). However, knockdown of RBFOX3 dramatically decreased the number of metastatic tumors in the lungs in contrast to the non-sense shRNA control, and overexpression of hTERT in the RBFOX3 knockdown Hep3B cell lines increased the number of metastatic tumors in the lungs compared to the vector control (Figure 4H). None of the tail vein injection in nude mice affected their body weight and health condition (data was not shown). Taken together, these results indicated that RBFOX3 regulated HCC cell migration and invasion via hTERT signaling pathway in vitro and in vivo.
RBFOX3 Regulated HCC Cell Apoptosis and Cell Cycle via hTERT Signaling Pathway
hTERT signaling pathway has been shown to participate in apoptosis. We also examined whether RBFOX3 could participate in apoptosis via hTERT signaling. The results showed that the number of apoptotic cells was increased in the RBFOX3-knocked down Hep3B and HepG2 cells (Figure 5A and Supplementary Figure 5A), and knockdown of RBFOX3 increased the expression of cleaved caspase 3, cleaved caspase 9, and cleaved PARP (89 kDa) (Figure 5B and Supplementary Figure 5B). Moreover, overexpression of hTERT rescued the cell apoptosis mediated by RBFOX3 knockdown (Figure 5C).
RBFOX3 regulated HCC cell apoptosis and cell cycle progression via hTERT signaling pathway. (A) The apoptotic cells were analyzed in the RBFOX3 knockdown Hep3B cells. Transfection reagent treated cells served as the mock control. (B) Expression of pro-apoptotic protein cleaved-caspase9, cleaved-caspase3, and anti-apoptotic protein PARP were detected and quantified in RBFOX3 knockdown Hep3B cells. The relative protein level was normalized to the expression of GAPDH. (C) Overexpression of hTERT reversed the induction of cell apoptosis by RBFOX3 knockdown in Hep3B cells. (D) The number of G0/G1 phase cells was decreased in RBFOX3 overexpressing QGY7703 cells compared with vector overexpressing QGY7703 cells. (E) The number of G0/G1 phase cells was increased in RBFOX3 knockdown Hep3B cells. (F) Overexpression of hTERT reversed the inhibition of cell cycle progression by RBFOX3 knockdown in Hep3B cells. (G) Expression of cell cycle related protein p27, Cyclin D1 and Cyclin A were detected and quantified in RBFOX3 overexpressing QGY7703 cells. (H) Expression of cell cycle related protein p27, Cyclin D1 and Cyclin A were detected and quantified in RBFOX3 knockdown Hep3B cells. Cell cycle and cell apoptosis were tested by fluorescence-activated cell sorting (FACS). Data were shown as means ± SD. *p<0.05, by two-tailed Student's t-test. The experiments were performed in at least 3 independent trials.
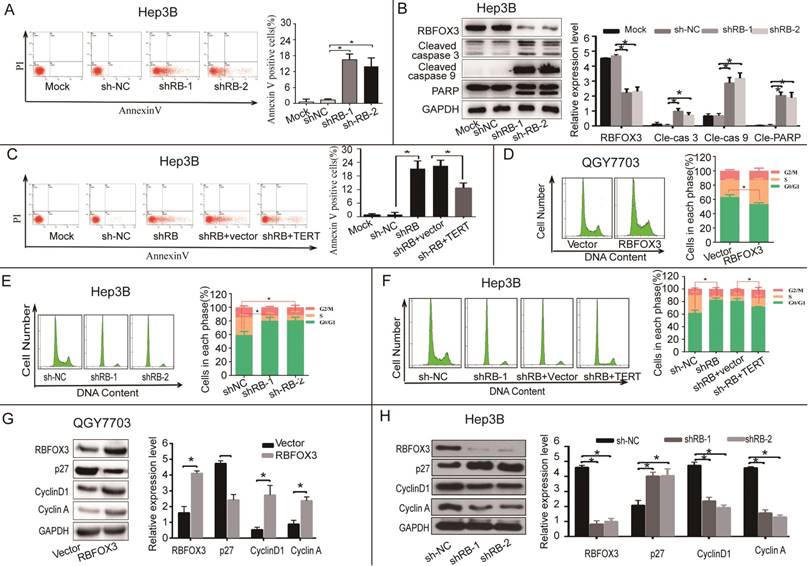
hTERT also has a vital function in regulating cell cycle progression. We next investigated whether RBFOX3 participated in HCC cell cycle regulation through the hTERT signaling pathway. We measured the cell number in each phase of mitosis in HCC cells in the conditions of RBFOX3 overexpressing and knockdown using FACS analysis. We found that the number of cells in G0/G1 phase was decreased in RBFOX3 overexpressing QGY7703 and SNU-449 cells (Figure 5D and Supplementary Figure 5C). However, the number of cells in G0/G1 phase was elevated markedly in RBFOX3 knockdown Hep3B and HepG2 cells compared to the non-sense shRNA control (Figure 5E and Supplementary Figure 5D). Moreover, we found that overexpression of hTERT in RBFOX3 knockdown Hep3B and QGY7703 cells rescued the effect of RBFOX3 knockdown in the progression of HCC cell cycle (Figure 5F and Supplementary Figure 5E).
We also examined the expression of cell cycle related proteins in cells with RBFOX3 knockdown or overexpression cells. We found that overexpression of RBFOX3 promoted cyclin D1 and cyclin A but inhibited p27 expression (Figure 5G and Supplementary Figure 5F), while knockdown of RBFOX3 promoted p27 but inhibited cyclin D1 and cyclin A expression (Figure 5H and Supplementary Figure 5G). These results indicated that RBFOX3 participated in cell cycle in HCC cells via hTERT signaling pathway.
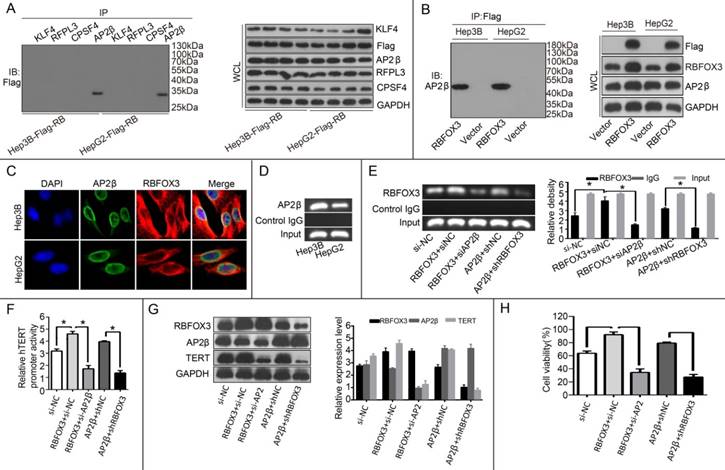
RBFOX3 interacted with AP-2β to regulate the hTERT expression
Our results above proved that RBFOX3 bound to the hTERT promoter and regulated the hTERT expression. The molecular mechanism by which RBFOX3 bound to the hTERT promoter was still unclear. Next, we tested whether RBFOX3 bound to the hTERT promoter by interacting with other transcription factors. To test this hypothesis, four hTERT promoter-binding proteins KLF4 [25], RFPL3 [26], CPSF4 [27] and AP-2β [23] previously identified were tested using their corresponding specific antibodies by a co-immunoprecipitation assay in the flag-RBFOX3 overexpressing Hep3B and HepG2 cells. Surprisingly, RBFOX3 could be pulled down by anti-AP-2β antibody (Figure 6A). The interaction between RBFOX3 and AP-2β was further proved by another co-immunoprecipitation assay with RBFOX3 specific antibody (Figure 6B). Moreover, we used dual immunofluorescence analysis to further analyze the expression and sub-cellular localization of RBFOX3 and AP-2β in Hep3B and HepG2 cells. As was shown in Figure 6C, RBFOX3 (red) was expressed both in nucleus and cytoplasm, and AP-2β (green) was primarily expressed in nucleus. The co-localization of AP-2β and RBFOX3 was observed at nucleus (yellow). The ChIP assay also confirmed the binding of AP-2β on the hTERT promoter (Figure 6D). Overexpression of AP-2β enhanced the binding of RBFOX3 on hTERT promoter (Figure 6E). On the contrary, AP-2β knockdown weakened the binding of RBFOX3 on hTERT promoter even in RBFOX3 overexpressing Hep3B cells (Figure 6E). The luciferase reporter assay also showed that overexpression of AP-2β enhanced the hTERT promoter activity while AP-2β knockdown inhibited the hTERT promoter activity (Figure 6F). To test the effect of AP-2β on the expression of hTERT, Western blot was performed. As shown in Figure 6G, overexpression of AP-2β up-regulated the expression of hTERT while hTERT expression was down-regulated with AP-2β knockdown. Moreover, we also tested the effect of AP-2β on cell viability by a MTS assay, and found that AP-2β knockdown inhibited the HCC cell growth increased by RBFOX3 overexpression while AP-2β overexpression partly rescued RBFOX3 knockdown-mediated growth inhibition. These results above indicated that RBFOX3 interacted with AP-2β to regulate the hTERT expression and growth in HCC cells.
RBFOX3 interacted with AP-2β and regulated the hTERT expression. (A) The extracted proteins from Hep3B and HepG2 cells with stable overexpression of flag-RBFOX3 were immunoprecipitated with antibody against KLF4 or RFPL3 or CPSF4 or AP-2β. The precipitates were analyzed by immunoblot using anti-flag antibody (left panel). The whole cell lysate (WCL) was analyzed by immunoblot (right panel). (B) The extracted proteins from Hep3B and HepG2 cells with stable overexpression of flag-RBFOX3 were immunoprecipitated with antibody against Flag. The precipitates were analyzed by immunoblot using anti-AP-2β antibody (left panel). The whole cell lysate (WCL) was analyzed by immunoblot (right panel). (C) Hep3B and HepG2 cells grown on chamber slides were cultivated for 24 h, and the subcellular localization and the colocalization of RBFOX3 with AP-2β were examined by confocal microscopy analysis. (D) Chromatin immunoprecipitation assays (ChIP) were done using antibody against AP-2β. The PCR products of hTERT promoter (-378 to -157) were separated on 2% agarose gels. (E) ChIP assays were carried out using the hTERT promoter primers and RBFOX3 antibody in Hep3B cells transfected with RBFOX3, RBFOX3 and siAP-2β, AP-2β, AP-2β and shRBFOX3, respectively. (F) Relative hTERT promoter activity in Hep3B cells transfected with RBFOX3, RBFOX3 and siAP-2β, AP-2β, AP-2β and shRBFOX3, respectively. (G) The expression of hTERT in Hep3B cells transfected with RBFOX3, RBFOX3 and siAP-2β, AP-2β, AP-2β and shRBFOX3, respectively. (H) MTS was performed in Hep3B cells transfected with RBFOX3, RBFOX3 and siAP-2β, AP-2β, AP-2β and shRBFOX3, respectively.
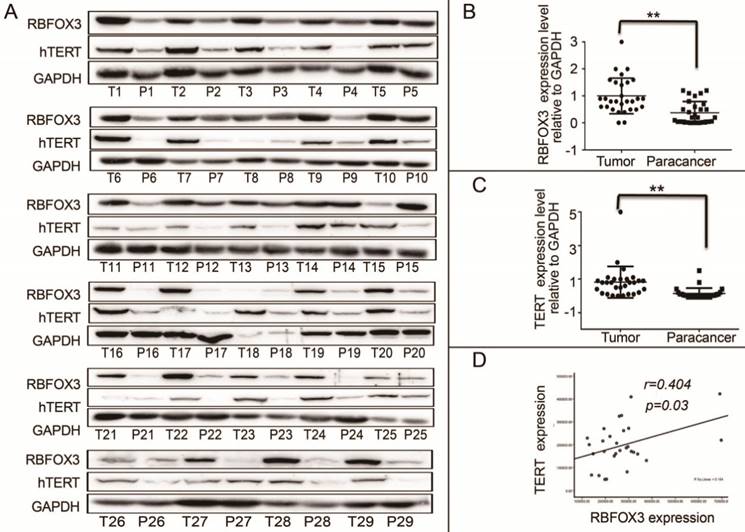
RBFOX3 and hTERT Were Positively Correlated and Highly Expressed in Tumor Tissues of HCC Patients
We demonstrated that RBFOX3 regulated hTERT expression via binding to hTERT promoter and promoting hTERT transcription. However, the relationship of RBFOX3 and hTERT expression in HCC patient samples was not investigated. We collected 30 (29/30 were available) HCC patient liver tumor tissues and the corresponding paracancer tissues. We found that the protein levels of RBFOX3 and hTERT were significantly higher in HCC tissues than the corresponding paracancer tissues (Figure 7A-C). Moreover, we found that the expression of RBFOX3 was positively correlated with the expression of hTERT (Pearson correlation test, n=29, r=0.404, p=0.03) (Figure 7D). These results further suggested the regulation of hTERT by RBFOX3 in HCC.
RBFOX3 and hTERT were correlated and highly expressed in tumor tissues of HCC patients. (A) RBFOX3 protein expression was analyzed by Western blot in 29 human HCC tissues (T) and the corresponding paracancer tissues samples (P) by Western blot analysis. T, HCC patient liver tumor tissues. P, the corresponding paracancer tissues. (B) RBFOX3 expression was analyzed in HCC samples and the corresponding paracancer tissues samples. (C) hTERT expression was analyzed in HCC samples and the corresponding paracancer tissues samples. (D) Correlation between RBFOX3 and hTERT protein levels in human HCC samples. Statistical test indicated a significant (**p<0.05) positive correlation, by two-tailed Pearson correlation test, n=29, r=0.404, p=0.03.
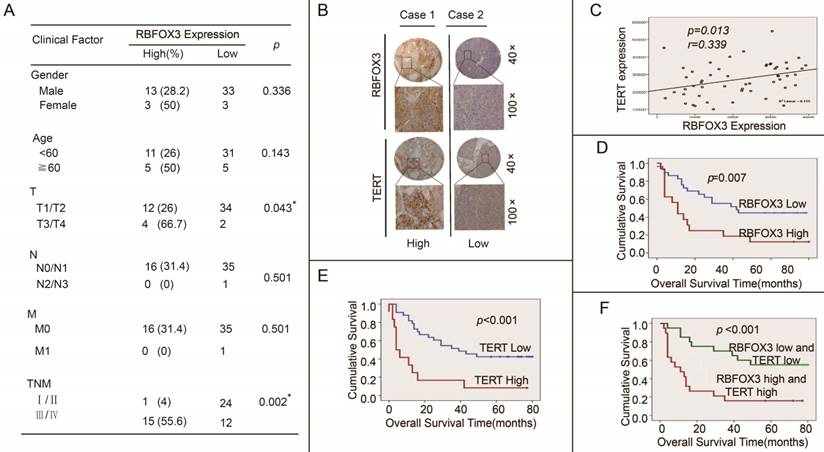
High RBFOX3 and hTERT Expression Was Associated with Poor Clinical Outcomes in HCC Patients
We further investigated the effect of high expression of RBFOX3 and hTERT on HCC patients' clinical outcomes using the tissue microarrays (n=52). The correlation between RBFOX3 expression and clinicopathologic variables of 52 HCC patients was shown in Figure 8A. We found that high expression of RBFOX3 was significantly associated with larger tumor (T, p=0.043) and later stage of TNM (TNM, p=0.002). No correlation was observed between RBFOX3 expression and patient gender (p=0.336), age (p=0.143), lymph node metastasis (N, p=0.501) and distant metastasis (M, p=0.501). We also found that RBFOX3 and hTERT were highly expressed in HCC tissues and positively correlated (Figure 8B and 8C). This result was consistent with Figure 7D. Furthermore, patients with high RBFOX3 expression had a shorter overall survival time than those with low RBFOX3 expression (Figure 8D), which was also the case in hTERT (Figure 8E). Additionally, the overall survival time of the patients who had high expression of both RBFOX3 and hTERT was much shorter than those whose RBFOX3 and hTERT were lowly expressed (Figure 8F). These results demonstrated that RBFOX3/hTERT was a key factor affecting HCC patients' clinical outcomes, and could be a potential therapeutic target in HCC.
High RBFOX3 and hTERT expression was associated with poor clinical outcomes in HCC patients. (A) Correlation analyses of RBFOX3 protein expression in relation to clinicopathologic variables of 52 HCC patients. (B) Two representative images of immunohistochemical analysis of RBFOX3 and hTERT protein level from HCC tissue microarray. “Low” indicted both RBFOX3 and hTERT had low expression and “high” indicted both RBFOX3 and hTERT had high expression. (C) Correlation between RBFOX3 expression and hTERT expression in 52 HCC paraffin section samples from HCC tissue microarray. Pearson correlation test, n=52, r=0.339, p=0.013. (D, E) Kaplan-Meier overall survival curves for HCC patients with low (blue line) or high (red line) expression of RBFOX3 (D) and hTERT (E). (F) The overall survival status of HCC patients with low expression of both hTERT and RBFOX3 and high expression of both hTERT and RBFOX3.
Taken together, our results demonstrated that RBFOX3 was an hTERT promoter-binding protein in HCC cells, and RBFOX3 promotes hTERT promoter activity and expression, thus promoting growth, migration, invasion and cell cycle progression in HCC cells (Graphical abstract). Finally, we found RBFOX3 and hTERT were correlated, highly expressed and associated with poor clinical outcomes in HCC patients.
Discussion
hTERT and telomerase RNA component (TERC) are the main components of telomerase, which adds telomere repeats to chromosome ends to offset the loss of telomere sequences that occurs due to the end-replication problem and is critical for telomere maintenance. As the catalytic subunit of telomerase, hTERT plays a decisive role in cell unlimited replication. In recent years, the roles of hTERT in human diseases, especially cancer, have attracted much attention. Currently, hTERT is thought to be a hallmark of cancer and a new target for cancer therapy [28]. Accumulating evidence has shown that tumor-specific cellular factors may be differentially expressed and specifically bind to the hTERT promoter to control hTERT expression and tumor development. The key findings of the current study are that RBFOX3, for the first time, was identified as an hTERT promoter-binding protein in hepatocellular carcinoma (HCC) cells, and that the hTERT promoter was activated when RBFOX3 bound to it. Our study also showed that hTERT expression was regulated at the transcriptional level by RBFOX3. We found that RBFOX3 expression was frequently up-regulated in primary human HCC cell lines (HepG2, Hep3B, QGY7703 and SNU-449) compared with the immortalized hepatocytes (LO2). Down-regulation of RBFOX3 suppressed HCC cell growth, migration, invasion, and cell cycle progression and induced cell apoptosis. Conversely, up-regulation of RBFOX3 promoted HCC cell growth, migration, invasion, and cell cycle progression. We also found that the inhibition of cell growth, cell cycle progression, cell migration and invasion and the induction of cell apoptosis mediated by RBFOX3 knockdown could be rescued by hTERT overexpression. In addition, the growth and progression enhancing effect of RBFOX3 overexpression was confirmed by in vivo tumor growth assays. We found RBFOX3 promoted xenograft tumor growth and metastatic tumor formation in the lungs via hTERT signaling pathway in vivo. Moreover, we found RBFOX3 interacted with AP-2β and regulated the hTERT expression. These results further demonstrated that RBFOX3 interacted with AP-2β and regulated HCC growth and progression via hTERT signaling pathway. Finally, our results showed that RBFOX3 and hTERT were consistently highly expressed in tumor tissues of HCC patients compared with the corresponding paracancer tissues. High expression of RBFOX3 or hTERT alone or both was correlated with disease progression and poor prognosis, suggesting that RBFOX3 might contribute to the susceptibility to HCC. RBFOX3 is an antigen of the neuronal marker antibody NeuN. The expression of RBFOX3 in the hepatocellular carcinoma is surprising and interesting. Previous reports regarding the involvement of RBFOX3 in the tumors are very few. Kim et al. found that the number of RBFOX3-positive cells in tumorous lung tissues was lower than that in normal lung tissues, and RBFOX3 expression was inhibited during TGF-β-induced EMT [29]. Another study, however, showed that RBFOX3 was frequently (72%) expressed in non-small cell lung cancer and provided evidence that stem marker Oct4 gave rise to cancer cells expressing RBFOX3 [30]. However, the role of RBFOX3 in cancers especially hepatocellular carcinoma is still unclear. Our results found the important regulatory relationships between RBFOX3 and hTERT, and how such relationship might open up a broader field for the study of RBFOX3 in human cancers.
The other two members of the fox-1 family, RBFOX1 and RBFOX2, have been reported to be involved in tumorigenesis [31], transcription regulation, epithelial to mesenchymal transition (EMT) [32], and pluripotent stem cells growth and differentiation [11,33]. However, neither RBFOX1 nor RBFOX2 could regulate the expression of hTERT at transcriptional and translational levels in HCC cells. As RBFOX3 is a well-recognized neuronal marker, we also investigated whether RBFOX3-mediated hTERT gene regulation occurred in the neuronal cells. We found that knockdown of RBFOX3 slightly down-regulated the expression of hTERT in U251 cell line. However, the change in hTERT expression is not obvious when RBFOX3 was knockdown in N9 MG, U373, and U138 cell lines. Furthermore, we found the binding of RBFOX3 to the hTERT promoter in vivo was much weaker in neuronal tumor cell lines than in HCC cells. According to these results, we speculate that some co-regulators that RBFOX3 depends on to bind the hTERT promoter in HCCs might have little or no expression in neuronal tumor cell lines, and hence we have indicated that RBFOX3 might not be the major regulator of hTERT in the neuronal tumor cells.
Recent studies on RBFOX3 have repeatedly suggested that RBFOX3 is an active biological molecule, rather than just an alternative splicing factor. A previous study showed that RBFOX3 could bind to DNA in vitro [21]. RBFOX3 was also found to be involved in transcriptional regulation and control the biogenesis of a subset of microRNAs [22]. Nonetheless, in spite of our increased knowledge in RBFOX3, its contribution to the development of mammalian cells and its functional characterization remain elusive. RBFOX3 contains an RNA recognition motif (RRM)-type RNA binding domain (RBD) and RBFOX3 regulates splicing of many transcripts by binding the sequence (U)GCAUG of RNA through this domain. Here, we have presented strong evidence that RBFOX3 binds to the promoter of hTERT and regulates its expression, and HCC cell growth. However, whether the RNA-binding activity of RBFOX3 through RRM domain is involved in its DNA binding activity is still unknown. Even so, our finding reveals a new role of RBFOX3 and it will be interesting to further examine the role of RBFOX3 in tumorigenesis in future studies.
Previous studies have pinpointed hTERT as a proto-oncogene that contributes to the development and progression of diverse cellular processes and signaling pathways in multiple types of tumors [34-37]. In our study, we found that RBFOX3 regulated HCC development and progression via hTERT downstream signaling pathways. We observed that overexpression of RBFOX3 up-regulated hTERT expression and then activated PI3K/AKT signaling, resulting in elevated cyclin A and cyclin D but decreased p27 expression, which promoted HCC cell cycle progression through the G1-S transition [38-44]. These are consistent with the findings from previous studies that knockdown of hTERT inactivated PI3K/AKT signaling and induced G1 phase-arrest in cells [45,46]. Accumulating evidence has indicated that MMPs are also involved in multiple pathological processes, including tumor invasion and metastases, in experimental cancer models and in human malignancies [47,48]. Prior work suggests that MMP2 and MMP9, two important members of MMPs, are downstream effectors of hTERT signaling. Overexpression of hTERT up-regulated the expression of MMP2 and MMP9, which contributed to cells migration and invasion [49]. Our results also showed that RBFOX3 regulated HCC cell migration and invasion in vitro and in vivo via hTERT/MMP2/MMP9 signal pathway. Another important effect downstream of hTERT is caspase 3/PARP-mediated apoptosis [44]. We found that knockdown of RBFOX3 in HCC cells elevated cleaved caspase 3, cleaved caspase 9 and cleaved PARP, consistent with previous finding that hTERT played an anti-apoptotic role in tumor development [50]. Taken together, these results suggest that RBFOX3 regulates HCC development and progression via hTERT-mediated downstream signaling pathways.
In summary, our study has illustrated that as an oncogene, RBFOX3 facilitates tumor cell proliferation and induces cell cycle progression in HCCs. Furthermore, we have found that RBFOX3 exerts its oncogenic effect through the up-regulation of hTERT signaling in HCCs. Our findings have provided new insights into the understanding of the regulation of hTERT in HCC tumorigenesis, revealing that RBFOX3 is a critical regulatory molecule in the hTERT signaling pathway for tumor progression, and is a potentially effective target for HCC therapy.
Acknowledgements
This work was supported by the funds from the National Natural Science Foundation of China (81472178 WD, 81322029 LK, 81402254 DS), the State “973 Program” of China (2014CB542005), the Natural Science Foundation of Guangdong Province (2016A03031100 WD, 2015A030313018 WD), and the foundation of Guangdong Esophageal Cancer Institute (2015A09 WD).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Marquardt JU, Thorgeirsson SS. SnapShot: Hepatocellular Carcinoma. Cancer Cell. 2014;25:550-1
2. Zhang D, Zheng A, Li J. et al. Smart Cu(II)-aptamer complexes based gold nanoplatform for tumor micro-environment triggered programmable intracellular prodrug release, photodynamic treatment and aggregation induced photothermal therapy of hepatocellular carcinoma. Theranostics. 2017;7:164-79
3. Torre LA, Bray F, Siegel RL. et al. Global Cancer Statistics, 2012. CA: a cancer journal of clinicians. 2015;65:87-108
4. Robinson PJ, Reddel RR. Protein Composition of Catalytically. Science. 2007;315:1850-3
5. Moyzis RK, Buckingham JM, Cram LS. et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. 1988;85:6622-6
6. Kim YH, Kim KT, Lee SJ. et al. Image-aided suicide gene therapy utilizing multifunctional hTERT-targeting adenovirus for clinical translation in hepatocellular carcinoma. Theranostics. 2016;6:357-68
7. Lingner J, Hughes TR, Shevchenko A. et al. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276:561-7
8. Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT). Gene. 2012;498:135-46
9. Colgin LM, Reddel RR. Telomere maintenance mechanisms and cellular immortalization. Curr Opin Genet Dev. 1999;9:97-103
10. Bryan TM, Englezou A, Dunham MA. et al. Telomere length dynamics in telomerase-positive immortal human cell populations. Exp Cell Res. 1998;239:370-8
11. Bodnar AG, Ouellette M, Frolkis M. et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349-52
12. Kim NW, Piatyszek MA, Prowse KR. et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011-5
13. Cong YS, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: Organization of the gene and characterization of the promoter. Hum Mol Genet. 1999;8:137-42
14. Duan W, Zhang YP, Hou Z. et al. Novel Insights into NeuN: from Neuronal Marker to Splicing Regulator. Mol Neurobiol. 2016;53:1637-47
15. Yeo GW, Coufal NG, Liang TY. et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130-7
16. Gallagher TL, Arribere JA, Geurts PA. et al. Rbfox-regulated alternative splicing is critical for zebrafish cardiac and skeletal muscle function. Dev Biol. 2011;359:251-61
17. Dredge BK, Jensen KB. NeuN/Rbfox3 Nuclear and Cytoplasmic isoforms differentially regulate alternative splicing and nonsense-mediated decay of Rbfox2. PLoS One. 2011;6:e21585
18. Kim KK, Kim YC, Adelstein RS. et al. Fox-3 and PSF interact to activate neural cell-specific alternative splicing. Nucleic Acids Res. 2011;39:3064-78
19. McManus CJ, Graveley BR. RNA structure and the mechanisms of alternative splicing. Curr Opin Genet Dev. 2011;21:373-9
20. Kim KK, Adelstein RS, Kawamoto S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem. 2009;284:31052-61
21. Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201-11
22. Kim KK, Yang Y, Zhu J. et al. Rbfox3 Controls the Biogenesis of a Subset of MicroRNAs. Nat Struct Mol Biol. 2014;21:901-10
23. Deng WG, Jayachandran G, Wu G. et al. Tumor-specific activation of human telomerase reverses transcriptase promoter activity by activating enhancer-binding protein-2beta in human lung cancer cells. J Biol Chem. 2007;282:26460-70
24. Deng WG, Tang ST, Tseng HP. et al. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood. 2006;108:518-24
25. Hu W, Jia Y, Yu Z. et al. KLF4 downregulates hTERT expression and telomerase activity to inhibit lung carcinoma growth. Oncotarget. 2016;7:52870-87
26. Chen W, Lu J, Qin Y. et al. Ret finger protein-like 3 promotes tumor cell growth by activating telomerase reverse transcriptase expression in human lung cancer cells. Oncotarget. 2014;5:11909-23
27. Chen W, Qin L, Wang S. et al. CPSF4 activates telomerase reverse transcriptase and predicts poor prognosis in human lung adenocarcinomas. Mol Oncol. 2014;8:704-16
28. Aschacher T, Wolf B, Enzmann F. et al. LINE-1 induces hTERT and ensures telomere maintenance in tumour cell lines. Oncogene. 2016;35:94-104
29. Kim YE, Kim JO, Park KS. et al. Transforming Growth Factor-beta-Induced RBFOX3 Inhibition Promotes Epithelial-Mesenchymal Transition of Lung Cancer Cells. Mol Cells. 2016;39:625-30
30. Langenfeld E, Deen M, Zachariah E. et al. Small molecule antagonist of the bone morphogenetic protein type I receptors suppresses growth and expression of Id1 and Id3 in lung cancer cells expressing Oct4 or nestin. Mol Cancer. 2013;12:129
31. Sengupta N, Yau C, Sakthianandeswaren A. et al. Analysis of colorectal cancers in British Bangladeshi identifies early onset, frequent mucinous histotype and a high prevalence of RBFOX1 deletion. Mol Cancer. 2013;12:1
32. Venables JP, Brosseau J-P, Gadea G. et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol. 2013;33:396-405
33. Venables JP, Lapasset L, Gadea G. et al. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat Commun. 2013;4:2480
34. Smith LL, Coller Ha, Roberts JM. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol. 2003;5:474-9
35. Choi J, Southworth LK, Sarin KY. et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008;4:e10
36. Koh CM, Khattar E, Leow SC. et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest. 2015;125:2109-22
37. Liu N, Ding D, Hao W. et al. hTERT promotes tumor angiogenesis by activating VEGF via interactions with the Sp1 transcription factor. Nucleic Acids Res. 2016;44:8693-8703
38. Lanza C, Tan EP, Zhang Z. et al. Reduced O-GlcNAcase expression promotes mitotic errors and spindle defects. Cell Cycle. 2016;15:1363-75
39. Mukherjee S, Chakraborty P, Saha P. Phosphorylation of Ku70 subunit by cell cycle kinases modulates the replication related function of Ku heterodimer. Nucleic Acids Res. 2016;44:7755-65
40. Wang Z, Ying M, Wu Q. et al. Overexpression of myosin VI regulates gastric cancer cell progression. Gene. 2016;593:100-9
41. Song Z, Fusco J, Zimmerman R. et al. EGFR signaling regulates beta cell proliferation in adult mice. J Biol Chem. 2016;291:22630-37
42. Cai F, Zhu Q, Miao Y. et al. Desmoglein-2 is overexpressed in non-small cell lung cancer tissues and its knockdown suppresses NSCLC growth by regulation of p27 and CDK2. J Cancer Res Clin Oncol. 2017;143:59-69
43. Zheng J, Huang X, Tan W. et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat Genet. 2016;48:747-57
44. Giunco S, Dolcetti R, Keppel S. et al. hTERT inhibition triggers Epstein-Barr virus lytic cycle and apoptosis in immortalized and transformed B cells: A basis for new therapies. Clin Cancer Res. 2013;19:2036-47
45. Li H, He J, Yi H. et al. siRNA suppression of hTERT using activatable cell-penetrating peptides in hepatoma cells. Biosci Rep. 2015;35:1-8
46. Maida Y, Kyo S, Kanaya T. et al. Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene. 2002;21:4071-9
47. Zhang Y, Gong LH, Zhang HQ. et al. Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett. 2010;293:189-97
48. Hee N, Gun D, Hee B. et al. Porphyromonas gingivalis increases the invasiveness of oral cancer cells by upregulating IL-8 and MMPs. Cytokine. 2016;86:64-72
49. Omi H, Okamoto A, Nikaido T. et al. Establishment of an immortalized human extravillous trophoblast cell line by retroviral infection of E6/E7/hTERT and its transcriptional profile during hypoxia and reoxygenation. Int J Mol Med. 2009;23:229-36
50. Jang KJ, Kwon GS, Jeong JW. et al. Cordyceptin induces apoptosis through repressing hTERT expression and inducing extranuclear export of hTERT. J Biosci Bioeng. 2015;119:351-7
Author contact
![]() Corresponding authors: Xiaojun Wu, Sun Yat-Sen University Cancer Center, Guangzhou 510060, China, E-mail: wuxjorg.cn; or Xinfa Yu, ShunDe Hospital of Southern Medical University, Guangdong 528300, E-mail: yuxfacom; or Wuguo Deng, Sun Yat-Sen University Cancer Center, Guangzhou 510060, China, E-mail: dengwgorg.cn
Corresponding authors: Xiaojun Wu, Sun Yat-Sen University Cancer Center, Guangzhou 510060, China, E-mail: wuxjorg.cn; or Xinfa Yu, ShunDe Hospital of Southern Medical University, Guangdong 528300, E-mail: yuxfacom; or Wuguo Deng, Sun Yat-Sen University Cancer Center, Guangzhou 510060, China, E-mail: dengwgorg.cn