Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(9):2329-2347. doi:10.7150/thno.21451 This issue Cite
Research Paper
Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy
Zheqiong Tan1,2,3, Lanbo Xiao1,2,3*, Min Tang1,2,3*, Fang Bai5, Jiangjiang Li1,2,3,4, Liling Li1,2,3, Feng Shi1,2,3, Namei Li1,2,3, Yueshuo Li1,2,3, Qianqian Du1,2,3, Jingchen Lu1,2,3, Xinxian Weng1,2,3, Wei Yi1,2,3, Hanwen Zhang6, Jia Fan7, Jian Zhou7, Qiang Gao7, José N. Onuchic5, Ann M. Bode8, Xiangjian Luo1,2,3 ![]() , Ya Cao1,2,3,4
, Ya Cao1,2,3,4 ![]()
1. Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China
2. Cancer Research Institute, Xiangya School of Medicine, Central South University, Changsha 410078, China
3. Key Laboratory of Carcinogenesis, Chinese Ministry of Health, Changsha 410078, China
4. Research Center for Technologies of Nucleic Acid-Based Diagnostics and Therapeutics Hunan Province, Changsha 410078, China
5. Center for Theoretical Biological Physics, Rice University, Houston, Texas 77005, USA
6. Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha 410078, China
7. Zhongshan Hospital and Shanghai Medical School, Fudan University, Key Laboratory for Carcinogenesis and Cancer Invasion, Chinese Ministry of Education, Shanghai 200000, China
8. The Hormel Institute, University of Minnesota, Austin, MN 55912, USA
*These authors contributed equally to this work.
Received 2017-6-13; Accepted 2018-2-28; Published 2018-3-22
Abstract

Nasopharyngeal carcinoma (NPC) has a particularly high prevalence in southern China, southeastern Asia and northern Africa. Radiation resistance remains a serious obstacle to successful treatment in NPC. This study aimed to explore the metabolic feature of radiation-resistant NPC cells and identify new molecular-targeted agents to improve the therapeutic effects of radiotherapy in NPC. Methods: Radiation-responsive and radiation-resistant NPC cells were used as the model system in vitro and in vivo. Metabolomics approach was used to illustrate the global metabolic changes. 13C isotopomer tracing experiment and Seahorse XF analysis were undertaken to determine the activity of fatty acid oxidation (FAO). qRT-PCR was performed to evaluate the expression of essential FAO genes including CPT1A. NPC tumor tissue microarray was used to investigate the prognostic role of CPT1A. Either RNA interference or pharmacological blockade by Etomoxir were used to inhibit CPT1A. Radiation resistance was evaluated by colony formation assay. Mitochondrial membrane potential, apoptosis and neutral lipid content were measured by flow cytometry analysis using JC-1, Annexin V and LipidTOX Red probe respectively. Molecular markers of mitochondrial apoptosis were detected by western blot. Xenografts were treated with Etomoxir, radiation, or a combination of Etomoxir and radiation. Mitochondrial apoptosis and lipid droplets content of tumor tissues were detected by cleaved caspase 9 and Oil Red O staining respectively. Liquid chromatography coupled with tandem mass spectrometry approach was used to identify CPT1A-binding proteins. The interaction of CPT1A and Rab14 were detected by immunoprecipitation, immunofluorescence and in situ proximity ligation analysis. Fragment docking and direct coupling combined computational protein-protein interaction prediction method were used to predict the binding interface. Fatty acid trafficking was measured by pulse-chase assay using BODIPY C16 and MitoTracker Red probe. Results: FAO was active in radiation-resistant NPC cells, and the rate-limiting enzyme of FAO, carnitine palmitoyl transferase 1 A (CPT1A), was consistently up-regulated in these cells. The protein level of CPT1A was significantly associated with poor overall survival of NPC patients following radiotherapy. Inhibition of CPT1A re-sensitized NPC cells to radiation therapy by activating mitochondrial apoptosis both in vitro and in vivo. In addition, we identified Rab14 as a novel CPT1A binding protein. The CPT1A-Rab14 interaction facilitated fatty acid trafficking from lipid droplets to mitochondria, which decreased radiation-induced lipid accumulation and maximized ATP production. Knockdown of Rab14 attenuated CPT1A-mediated fatty acid trafficking and radiation resistance. Conclusion: An active FAO is a vital signature of NPC radiation resistance. Targeting CPT1A could be a beneficial regimen to improve the therapeutic effects of radiotherapy in NPC patients. Importantly, the CPT1A-Rab14 interaction plays roles in CPT1A-mediated radiation resistance by facilitating fatty acid trafficking. This interaction could be an attractive interface for the discovery of novel CPT1A inhibitors.
Keywords: CPT1A, fatty acid oxidation, nasopharyngeal carcinoma, radiation therapy, Rab14, fatty acid trafficking
Introduction
Nasopharyngeal carcinoma (NPC) has a particularly high prevalence in southern China, southeastern Asia and northern Africa [1]. Due to the anatomic constraints for surgery, intensity-modulated radiotherapy is the primary treatment for non-disseminated NPC [2]. However, more than 70% of newly diagnosed NPC cases are classified as the advanced stage (stage III or IV). Radioresistance exists in some of these cases and can cause local recurrence and distant metastases [3, 4]. Chemotherapeutic drugs, such as 5-fluorouracil and platinum analogs, are commonly used in order to improve the efficacy of radiotherapy [5, 6]. However, several phase 3 studies reported that induction chemotherapy does not achieve better therapeutic effects than radiotherapy alone [7-9]. In addition, to develop truly effective induction chemotherapy drugs, identification of new molecular targeted agents to eliminate radiation resistance is also necessary.
In our previous study, a metabolomics-based discrimination model for NPC illustrated that fatty acids significantly contribute to the discrimination between serum from NPC patients and serum from healthy controls [10]. Additionally, the serum levels of free-linoleic acid and free-arachidonic acid, which were much lower in NPC patients at the diagnosed stage, gradually increased to normal after radiation therapy in about one year. These changes suggest that fatty acid metabolism is deregulated and associated with radiation therapy resistance in NPC.
Cancer cells depend on alternative metabolic pathways to acquire nutrients and produce building blocks or energy for survival [11-13]. In addition to the “Warburg effect”, fatty acids, glutamine, one-carbon unit metabolism and mitochondrial oxidative phosphorylation (Oxphos) are considered important participants in tumor progression [14-21]. Fatty acids are fundamental cellular components and energy sources. They are catabolized primarily by fatty acid oxidation (FAO), which produces more than three-fold as much ATP per mole as oxidation of carbohydrates [22]. FAO is relatively more active in energy-consuming tissues such as muscle tissues and some tumor tissues [23-28]. Recent evidence underscores the idea that FAO is increasingly recognized as a crucial metabolic characteristic of cancer [29, 30]. FAO replenishes the tricarboxylic acid (TCA) cycle and promotes NADPH and ATP production to overcome stress in cancer cells [31, 32]. Importantly, FAO is considered to be a subtype-specific metabolic signature in several cancer cell types, such as CD36+ leukemic stem cells, MYC-overexpressing triple-negative breast cancer and Oxphos-active diffuse large B-cell lymphoma [33-36]. Inhibition of FAO suppresses tumor growth in these cells, which indicates that targeting FAO might be a promising strategy in cancer therapy.
Carnitine palmitoyl transferase 1 (CPT1) controls the rate-limiting step of FAO. It facilitates the entry of fatty acids into the mitochondria by loading fatty acyl-groups onto carnitine. The CPT1 family comprises three subtypes, CPT1A (liver type), CPT1B (muscle type) and CPT1C (brain type) [37]. Recently, the mRNA level of CPT1C was found to be upregulated in non-small-cell lung cancer [31] and CPT1A expression was found to be related to a poor overall survival of ovarian cancer patients [38]. The evidence indicated that CPT1 might be a key metabolic target in cancer.
In the present study, using a metabolomics approach, we illustrated a metabolic feature of active lipid turnover and FAO in radiation-resistant NPC cells. High expression of CPT1A was confirmed to promote radiation resistance in NPC cells and contribute to a poor overall survival of NPC patients following radiation therapy. Disruption of CPT1A decreases radiation resistance by activating mitochondrial apoptosis both in vitro and in vivo. Interestingly, CPT1A binds to Rab14 and facilitates fatty acid trafficking between lipid droplets and mitochondria, which promotes fatty acid utilization and maximizes ATP production in NPC. Knockdown of Rab14 blunts CPT1A-mediated lipid metabolism and radiation resistance. Our study provides novel insights into the mechanism of CPT1A involvement in lipid reprogramming, and suggests new strategies of molecular-targeted treatment to improve the therapeutic effects of radiotherapy in NPC.
Materials and Methods
Cell lines and culture conditions
The radiation-resistant NPC cells, CNE2-IR and HK1-IR, were established as described [39-42]. Cells were irradiated at a dose rate of 2 Gy/min using the X-RAD255 (Precision X-ray, North Branford, CT). The NPC cell lines were cultured in RPMI-1640 medium (Hyclone, UT, USA) with 10% fetal bovine serum (FBS, Hyclone). The human embryonic kidney cell line HEK293T was cultured in Dulbecco's modified eagle medium (Hyclone) with 10% FBS (Hyclone). Cells were cultured at 37 °C in a 5% CO2 incubator.
Metabolic profiling
CNE2-IR and CNE2 cells were collected and quickly frozen. Further sample preparation, metabolic profiling, peak identification and curation were performed by Metabolon (Durham, NC, STATE, USA) [43].
Seahorse XF-24 metabolic flux analysis
At 24 h before the experiment, cells were cultured on XF-24 plates at a density of 3 × 104 cells/well. At 6 h before metabolic flux analysis, the culture medium was replaced with 600 μL of substrate-limited medium. Before the analysis, the medium was replaced with 410 μL FAO Assay Medium and incubated at 37°C in a non-CO2 incubator for 45 min. During the experiment, palmitate-BSA, BSA, and inhibitors were sequentially injected: palmitate-BSA (175 μM), BSA (175 μM), Etomoxir (80 μM), antimycin A (0.2 μM) and rotenone (0.2 μM). Then OCR was automatically calculated by the Seahorse XF-24 analyzer (Seahorse Bioscience, CA, USA).
Isotope tracing experiments
Cells (2 ×106) were incubated for 8 h at 37°C in RPMI 1640 medium with 10% fetal bovine serum (FBS). U-13C-palmitate (Sigma-Aldrich) was dissolved in ethanol to a final concentration of 20 mM, then mixed with a 10% free fatty acid-free bovine serum albumin (Sigma-Aldrich) solution at a 1:5 ratio and incubated 1 h at 37°C. Then the U-13C-palmitate solution was diluted in serum-containing RPMI 1640 medium to a working concentration of 50 μM. When tracing palmitate carbons, the cells were incubated with the medium indicated above. After 24 h, cells were quickly washed with 1×PBS and fixed with a pre-cooling methanol (HPLC-MS grade, Millipore) for 30 min at -80°C. Then cells were harvested, frozen on dry ice and processed for UPLC-MS/MS analysis as described below.
Plasmids and siRNA
Target sequences from the RNA interference Consortium shRNA Library were used to construct shRNA-targeting CPT1A. The specific sequences were cloned into the GV248 vector (Genechem, Shanghai, China). The empty vector PLXSN-V5 and PLXSN-CPT1A-V5 were purchased from Addgene (MA, USA). The human Rab14 siRNA smart pool and non-targeting pool were from Dharmacon (Cambridge, UK).
Western blot analysis and antibodies
Cells were harvested and disrupted in IP lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP40, 1 mM EDTA, 5% glycerol; Thermo Scientific, MA, USA). Extracted proteins were separated by SDS-PAGE and transferred onto nylon membranes. Binding of primary antibodies was detected using peroxidase-conjugated secondary antibodies. Visualization was performed using the ChemiDoc XRS system with Image Lab software (Bio-Rad, CA, USA). Antibodies included anti-CPT1A, anti-cleaved caspase 9, anti-MLKL (phospho S358) (Abcam, MA, USA), anti-cleaved-PARP, anti-cleaved caspase 3, anti-normal rabbit IgG and anti-normal mouse IgG (Cell Signaling Technologies, MA, USA), anti-Rab14, anti-Rab7A, anti-MLKL and anti-β-actin (Sigma-Aldrich, Darmstadt, Germany), anti-HSP60 (Santa Cruz, CA, USA), and anti-LC3 I/II (Novus Biologicals, CA, USA).
CPT1 enzymatic activity
CPT1 enzymatic activity was detected by measuring the release of CoA-SH from palmitoyl-CoA using the general thiol reagent DTNB [44-46]. The DTNB buffer (116 mM Tris-HCl pH 8.0, 0.09% Triton X-100, 1.1 mM EDTA, 0.12 mM DNTB) and cell lysate were mixed and incubated at room temperature for 30 min. The absorbance of buffer at 405 nm was defined as background. Then 100 μM palmitoyl-CoA and 5 mM L-carnitine were added to the mixture. After a 20 min incubation at 37°C, the absorbance was measured at 405 nm. The difference between readings with and without substrates was normalized to the Bradford protein concentration. CPT1 enzymatic activity was defined as millimoles of CoA-SH released per milligram of protein.
Co-immunoprecipitation
Cells were disrupted with IP lysis buffer containing protease inhibitor cocktails (Roche Diagnostics, Basel, Switzerland). Protein aliquots (500 μg) were pre-cleared by incubation with 20 μL of Dynabeads protein A (Invitrogen, MA, USA) for 1 h at 4 °C. The pre-cleared samples were incubated with antibody (2 μg/sample) overnight at 4 °C. Then 20 μL Dynabeads protein A were added to samples and incubated for 2 h at 4 °C. The beads were washed 3 times with cold lysis buffer, then boiled and analyzed by Western blot.
Proximity ligation assay
The DuoLink® In Situ Red Starter Kit Mouse/Rabbit (DUO92101, Sigma-Aldrich, Darmstadt, Germany) was used to detect interacting proteins. Cells were seeded in eight-well chamber slides (Millicell EZ SLIDE, Millipore, Darmstadt, Germany) and cultured overnight. Slides were washed with cold 1×PBS and fixed in 4% paraformaldehyde for 30 min. Then slides were blocked with Duolink Blocking Solution in a pre-heated humidified chamber for 30 min at 37°C. The primary antibody to detect CPT1A and Rab14 was added to the slides and incubated overnight at 4°C. Then slides were washed with 1×Wash Buffer A and subsequently incubated with the two PLA probes (1:5 diluted in antibody diluents) for 1 h, then the Ligation-Ligase solution for 30 min, and the Amplification-Polymerase solution for 100 min in a pre-heated humidified chamber at 37°C. Before imaging, slides were washed with 1×Wash Buffer B and mounted with a cover slip using Duolink In Situ Mounting Medium with DAPI. Fluorescence images were acquired using a Leica TCS SP8 confocal microscope.
Fluorescent fatty acid pulse-chase assay
NPC cells were treated with a 4 Gy dose of radiation, then incubated with complete medium containing 1 μM BODIPY C16 (Invitrogen, MA, USA) for 10 h. Cells were washed with complete medium and incubated for 1 h to allow the fluorescent fatty acids to metabolize. Then cells were chased for the time indicated (0 and 12 h). Mitochondria were labeled with 50 nM MitoTracker Red CMXRos (Invitrogen) for 30 min prior to imaging.
Immunofluorescence analysis
Mitochondria, lipid droplets and fatty acids were labeled with complete medium containing 50 nM MitoTracker Red CMXRos (Invitrogen), 1 μM BODIPY 493/503 (Invitrogen), or 1 μM BODIPY C16 (Invitrogen) for 30 min, respectively. Cells were washed with complete medium, and fixed with freshly prepared pre-warmed 1640 medium containing 3.7% formaldehyde at 37 °C for 15 min. Then the cells were rinsed with 1640 medium. Cells were blocked in 5% donkey serum in 1×PBS for 1 h at room temperature and incubated with a primary antibody in 1×PBS containing 1% BSA at 4 °C overnight. Cells were washed with 1×PBS and incubated with a secondary antibody for 45 min. Then cells were washed and stained with DAPI for 10 min and viewed by a confocal microscope (LSM 510 META, Germany).
Immunohistochemistry analysis
The NPC tissue array was purchased from Pantomics (Richmond, CA, USA). IHC was performed as previously described [47]. Images of the sections were acquired and scores of each section were quantified by two pathologists (Dr Liling Li and Dr Zhijie Xu, Xiangya Hospital, Changsha, China).
Colony formation and radiosensitivity assays
For Figures 2D-F, 4D, 6A, and Figures S3F, S4D-E, S7C, NPC cells were seeded at 1 × 103 cells/well in six-well plates in triplicate. For Figures S2A-B, NPC cells were seeded in six-well plates in triplicate at densities of 100, 400, 1 × 103 and 1 × 104 per well and were correspondingly exposed to 0, 2, 4 or 6 Gy IR. After 10-14 days, cells were washed with 1×PBS, fixed in methanol for 20 min and stained with crystal violet for 15 min at room temperature. Colonies containing more than 50 cells were counted using the Image J software and the survival fractions were calculated. The survival curves in Figure S2B were drawn using the GraphPad Prism 5 software program (GraphPad Software, La Jolla, CA, USA).
Xenograft studies
Animal experiments were conducted according to protocols approved by Xiangya Hospital (Changsha, China). CNE2-IR cells (5 × 106) were injected into the subcutaneous tissue over the right flank region of nude mice (BCLB/c-nu, male, 4 weeks old). Tumors grew to an average volume of 130 mm3 prior to initiation of therapy. CNE2-IR xenograft-bearing mice were randomly assigned into four groups (n = 4) as follows: (1) vehicle control (0.9% saline buffer, 100 μL, i.p., every other day for 3 weeks); (2) radiation (2 Gy, twice per week for 2 weeks) was delivered to the tumor; (3) Etomoxir (50 mg/kg, 100 μL, i.p., every other day for 3 weeks); (4) Etomoxir (50 mg/kg, 100 μL, i.p., every other day for 3 weeks) plus ionizing radiation (2 Gy, twice per week for two weeks, 2 h after ETO injection). Tumor volume was assessed by caliper measurements every other day and calculated according to the formula: V = (L x W2)/2. At the end of experiments, mice were euthanized by CO2 inhalation.
Statistics
The Kaplan-Meier method was used to estimate overall survival and the log-rank test was used to evaluate differences between survival curves. Statistical analyses were performed using the Student t-test or Welch's t-test (normal distribution). A p-value of < 0.05 was considered statistically significant. GraphPad Prism 5 software program (GraphPad Software, La Jolla, CA, USA) was used in most of the statistical analyses.
Study approval
The study was approved by the Ethics Committee of Xiangya Hospital. All procedures involving animal care and use were approved by the Institutional Animal Care and Usage Committee of Central South University, and were in accordance with the National Policy on Use of Laboratory Animals.
Results
Radiation-resistant NPC cells show a metabolic signature of active FAO
We used a metabolomics approach to profile the global metabolic changes of radiation-responsive (CNE2) and radiation-resistant (CNE2-IR) NPC cells. The contents of 260 biochemical compounds were quantified (Figure 1A), and we found obvious differences between the metabolic profiles of CNE2 and CNE2-IR cells. Relative to CNE2 cells, 130 biochemical factors were altered in CNE2-IR, and the general changes are shown as a pie chart (Figure 1B). Among these changes, 54 metabolic factors belong to lipid metabolism (Figure 1C). The phosphoglycerides degradation products, glycerol 3-phosphate, glycerolphosphorylcholine and multiple lysolipids, as well as the membrane sphingolipid metabolites, palmitoyl sphingomyelin and stearoyl sphingomyelin, were elevated in CNE2-IR cells (Figure S1A-C). These observations may reflect an increased lipid turnover or active lipid breakdown in CNE2-IR cells.
Lipid turnover and fatty acid oxidation are enhanced in radiation-resistant NPC cells. (A) Heat map of 260 biochemical factors in lysates from CNE2 and CNE2-IR cells. Five replicates were tested for each cell line. The relative fold change for each factor in each sample is represented as the relative average increase (red) or decrease (green). (B) Pie charts indicating the number of biochemical changes of each metabolic pathway in CNE2-IR cells compared with CNE2 cells (*p < 0.05, Welch's t-test). (C) Heat map of lipid factors in lysates from CNE2 and CNE2-IR cells. Five replicates were tested for each cell line. (D) Fold change of cellular acylcarnitine levels in CNE2-IR cells compared with CNE2 cells. Five replicates were tested for each cell line (*p < 0.05, **p < 0.01, ***p < 0.001). (E) Labeling incorporation from 13C16-palmitate into palmitoyl-carnitine (C16), myristoyl-carnitine (C14) and lauroyl-carnitine (C12) in CNE2-IR and HK1-IR cells compared with parental cells at 24 h after exposure to 4 Gy irradiation (IR). Data are shown as percentage of 13C16-C16, 13C14-C14, or 13C12-C12 to the total pool of each corresponding acylcarnitine. Three replicates were tested for each cell line (**p < 0.01, ***p < 0.001). (F) Oxygen consumption rates (OCR) were measured by Seahorse XF analysis with the indicated reagents in CNE2-IR and HK1-IR cells compared with parental cells at 24 h after exposure to 0 or 4 Gy IR. Arrows indicate the time when palmitate-BSA (175 μM), Etomoxir (80 μM), and antimycin (0.2 μM)/rotenone (0.2 μM) was added to cells (n = 3). BSA was used as a control for palmitate.
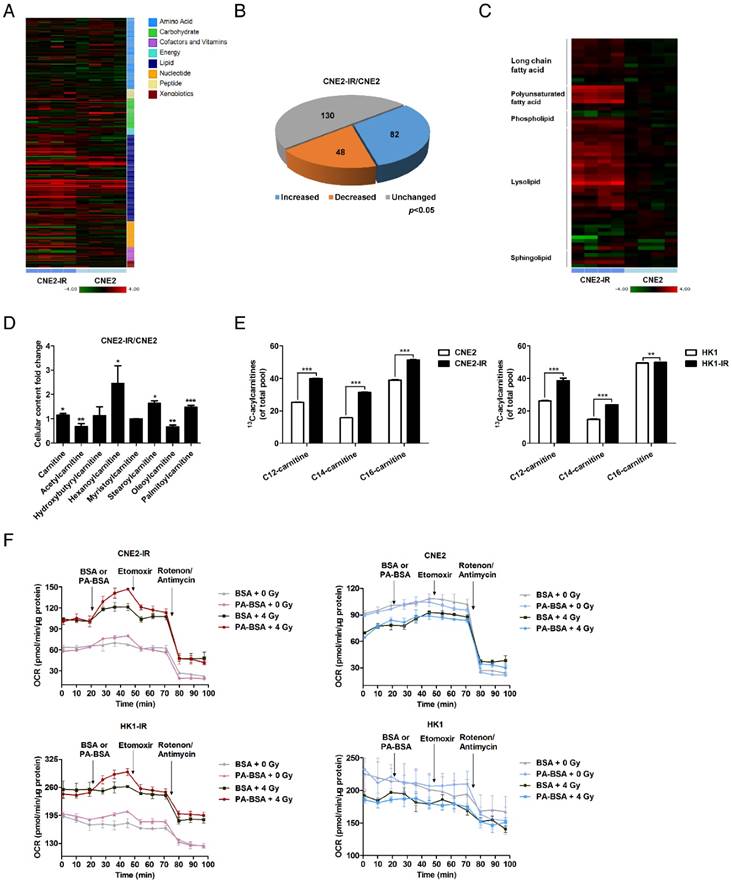
More importantly, we found that several acylcarnitine levels are substantially higher in CNE2-IR cells (Figure 1D). During the oxidation of fatty acids, carnitine acts as an acyl-CoA carrier facilitating the movement of fatty acids into the matrices of the mitochondria. To further explore the utilization of fatty acids through the carnitine shuttle system, a 13C isotopomer tracing experiment was undertaken. Cells were incubated with uniformly labeled 13C16-palmitate, and the abundance of 13C-labeled palmitoyl-carnitine (C16), myristoyl-carnitine (C14) and lauroyl-carnitine (C12) were measured by ultra-high-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS; Figure 1E). We found that 13C enrichment in acylcarnitines was higher in CNE2-IR and HK1-IR cells in response to a dose of 4 Gy radiation, indicating that radiation-resistant cells utilize palmitate and may have an active FAO. To determine the activity of FAO, we examined the palmitate (PA)-based oxygen consumption rate (OCR). Following radiation treatment, an obvious increase in OCR was observed in radiation-resistant cells after PA stimulation, which was then inhibited by ethyl 2-[6-(4-chlorophenoxy) hexyl] oxirane-2-carboxylate (Etomoxir, ETO), a specific FAO inhibitor [48] (Figure 1F and Figure S1D). In contrast, neither PA nor ETO treatment affected the OCR of radiation-responsive cells. These results in combination with the metabolomics analysis indicate an enhanced lipid turnover and FAO in radiation-resistant NPC cells, which suggests that lipid reprogramming might play a crucial role in mediating radiation resistance.
Radiation-resistant NPC cells depend on CPT1A to survive radiation therapy
To investigate the mechanisms of gain-of-FAO in radiation-resistant NPC cells, qRT-PCR was performed to evaluate the expression of essential FAO genes in NPC cells (Table S1). Several FAO genes are up-regulated in radiation-resistant cells (Figure 2A). Among these genes, CPT1A is most substantially highly transcribed. Its protein expression levels and enzyme activity are also increased in radiation-resistant cells, whether combined with 4 Gy of radiation or not (Figure 2B-C). ETO, a clinically tested, specific inhibitor of CPT1 was used to block the enzymatic activity of CPT1 (Figure 2C) [49]. We used a colony formation assay to evaluate radiation resistance of cells. To identify the radiation-resistant phenotypes, CNE2-IR and HK1-IR cells and their parental cells were exposed to different doses of IR (0, 2, 4, 6 Gy). The survival fractions indicate that radiation-resistant cells are authentically more tolerant to radiation treatment than radiation-responsive cells (Figure S2A-B). Cisplatin is a radiosensitizing agent widely applied in clinical therapy for NPC [50, 51]. Here we used it as a positive control to assess the effects of ETO. Survival rates and IC50 values were calculated (Figure S2C). The IC50 values of cisplatin were similar in each cell line, but the IC50 values of ETO in radiation-resistant cells were much lower compared with parental cells. Based on the data from radiation-resistant cells, we chose 50% of the IC50 value of each drug as the concentration for treatment in the following experiments. The results of colony formation showed that ETO markedly decreased the survival fraction of radiation-resistant cells, but had no effect on parental cells (Figure 2D and Figure S2D). In addition, cisplatin decreased the survival fraction of all the cell lines, but radiation-resistant cells are apparently more tolerant to this drug. Intriguingly, ETO-radiation combination achieved better inhibitory effects compared to the cisplatin-radiation combination.
CPT1A is up-regulated in radiation-resistant NPC cells and inhibition of CPT1A sensitizes these cells to radiation therapy. (A) Real-time PCR showing mRNA levels of FAO genes in CNE2-IR and HK1-IR cells compared with parental cells. Data are represented as mean values ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001). (B) Immunoblot analysis of CPT1A protein expression levels in indicated cells at 24 h after exposure to 0 or 4 Gy IR. β-Actin was used as a control to confirm equal loading of protein. (C) CPT1 enzymatic activity in lysates of indicated cells at 24 h after exposure to 0 or 4 Gy IR and Etomoxir (80 μM). Results are presented as fold change in enzymatic activity relative to each parental cell type. Data are represented as mean values ± S.D. of 3 independent experiments (***p < 0.001). (D) Colony formation assay showing survival fractions of the indicated cells following treatment with 0 or 4 Gy IR and Etomoxir (80 μM) or cisplatin (0.8 μM). Surviving fractions were calculated by comparing the colony number of each treatment group with untreated groups. Results are plotted as average survival fraction ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001). (E) Colony formation assay showing survival fractions of CNE2-IR cells stably transfected with CPT1A shRNA or control shRNA following treatment with 0 or 4 Gy IR. Survival fractions were calculated by comparing the colony number of each treatment group with untreated control shRNA group. Results are plotted as the average survival fraction ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001). (F) Colony formation assay showing survival fractions of CNE2 cells stably expressing CPT1A or empty vector following treatment with 0 or 4 Gy IR. Results are plotted as the average survival fraction ± S.D. of 3 independent experiments (**p < 0.01, ***p < 0.001).
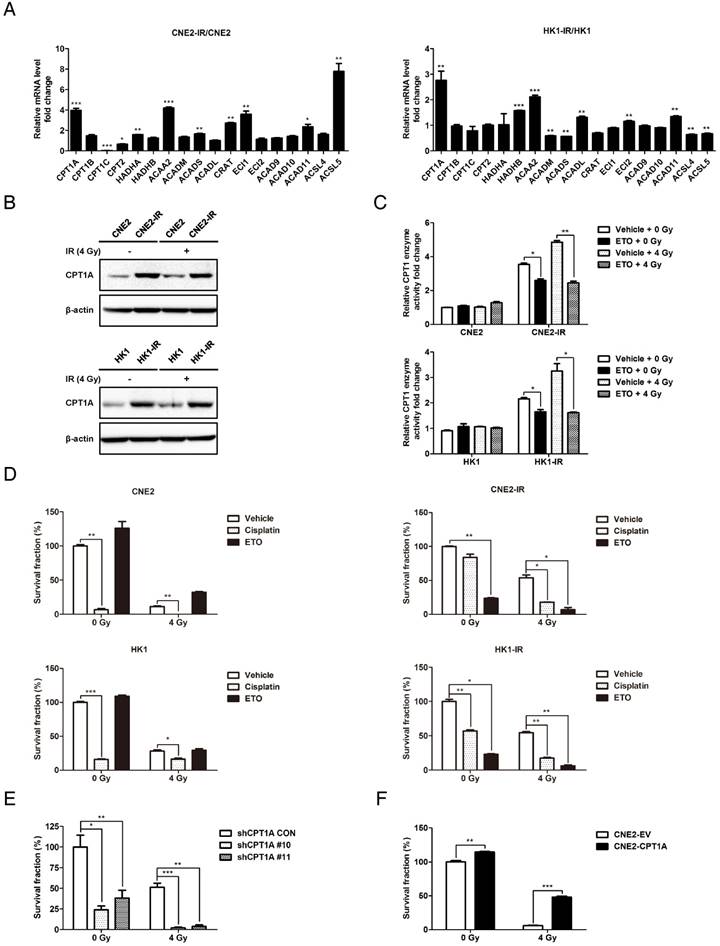
Furthermore, stable CPT1A knockdown CNE2-IR cells were constructed using a lentiviral-based CPT1A small hairpin RNA (shRNA; Figure S3A). To identify the effect of the loss of CPT1A on FAO, we examined the abundance of 13C-labeled acylcarnitines and PA-based OCR following radiation treatment. We observed a significant decrease in the 13C-labelling of acylcarnitines and PA-based OCR in CPT1A knockdown cells. These data indicate that knockdown of CPT1A reduces FAO in radiation-resistant cells (Figure S3B-C). Consistent with the effects of Etomoxir on radiation-resistant cells, the survival rate was obviously decreased by knockdown of CPT1A (Figure 2E and Figure S3D). On the other hand, CPT1A overexpression promotes radiation resistance in NPC cells (Figure 2F and Figure S3E-F). These data indicate that CPT1A is highly expressed in radiation-resistant NPC cells and is critical for these cells to survive radiation therapy.
Up-regulation of CPT1A corresponds with poor overall survival of NPC patients following radiation therapy
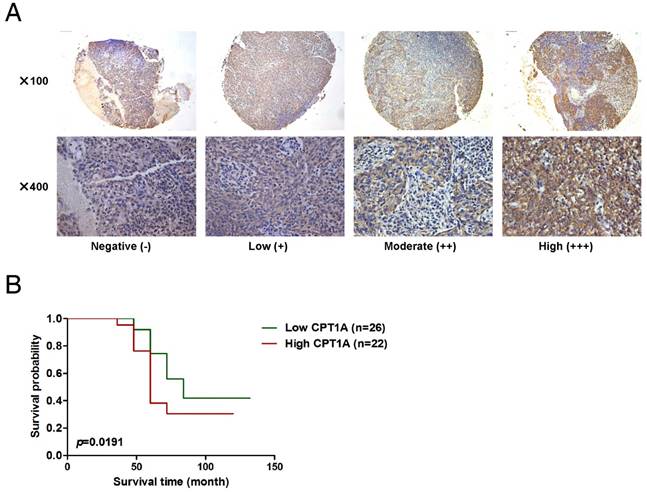
To investigate the prognostic role of CPT1A, a commercial NPC tumor tissue microarray was used (IHC, Figure 3A). Forty-eight non-keratinizing undifferentiated NPC patients who received radiation therapy were successfully followed-up. All of the tumor biopsies were obtained before treatment and patients' clinical characteristics are listed in Table S2. According to the IHC scores for CPT1A, we set the median score as the cut-off value and divided patients into two groups. The patients with high expression of CPT1A (n = 22, median survival time = 60 months) had shorter overall survival compared to patients expressing lower levels of CPT1A (n = 26, median survival time = 84 months; Figure 3B). These observations suggest that CPT1A overexpression is associated with a worse clinical outcome of radiation therapy in NPC.
Up-regulation of CPT1A corresponds with poor overall survival of NPC patients following radiation therapy. (A) Representative IHC staining of CPT1A expression from a tissue microarray of nasopharyngeal squamous cell carcinoma patients following radiation therapy. Scale bar: upper = 100 μm, lower = 25 μm. (B) Overall survival rates of nasopharyngeal squamous cell carcinoma patients with low (n = 26) or high (n = 22) expression levels of CPT1A were estimated with the Kaplan-Meier method by log-rank test following radiation therapy. The Kaplan-Meier curves were drawn using the GraphPad Prism 5 software program.
CPT1A is required to block mitochondrial apoptosis in radiation-resistant NPC cells
Various stress signals, such as damage triggered by irradiation, initiate mitochondrial apoptosis in cells. To gain insight into the role of CPT1A in mitochondrial apoptosis, we assessed the mitochondrial membrane potential (MMP) and apoptotic rate in NPC cells. The results show that radiation-resistant cells have a higher level of MMP, which could be decreased by ETO following radiation (Figure 4A). Radiation-resistant cells are more resistant to radiation-induced apoptosis (Figure 4B). However, ETO significantly activated apoptosis in these cells, which could be rescued by the caspase-inhibitor zVAD following radiation treatment (Figure 4B). Similar to these results, the levels of cleaved caspase 9, cleaved caspase 3, molecular markers of mitochondrial apoptosis, and cleaved poly ADP ribose polymerase (PARP) were markedly enhanced by ETO treatment, which could be alleviated by zVAD in response to radiation (Figure 4C). These results suggest that inhibition of CPT1A activates mitochondrial apoptosis in radiation-resistant cells.
Inhibition of CPT1A decreases radiation resistance by activating mitochondrial apoptosis, which can be recovered by treatment with zVAD in radiation-resistant NPC cells. (A) Flow cytometry analysis of MMP using JC-1 in CNE2-IR and HK1-IR cells compared with parental cells at 24 h after exposure to 4 Gy IR or Etomoxir (80 μM). Data represent JC-1 intensity mean values ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01). (B) Flow cytometry analysis of apoptosis using Annexin V in the cells indicated in (A) at 48 h after exposure to 4 Gy IR or Etomoxir (80 μM). Values represent Annexin V intensity mean values ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01). (C) Immunoblot analysis showing the expression levels of CPT1A, cleaved PARP, cleaved caspase 9 and cleaved caspase 3 in the groups indicated in (B). β-Actin was used as a control to confirm equal loading of protein. (D) Colony formation assay showing survival fractions of indicated cells treated with 4 Gy IR, Etomoxir (80 μM) or zVAD (20 μM). Results are plotted as the average survival fraction ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
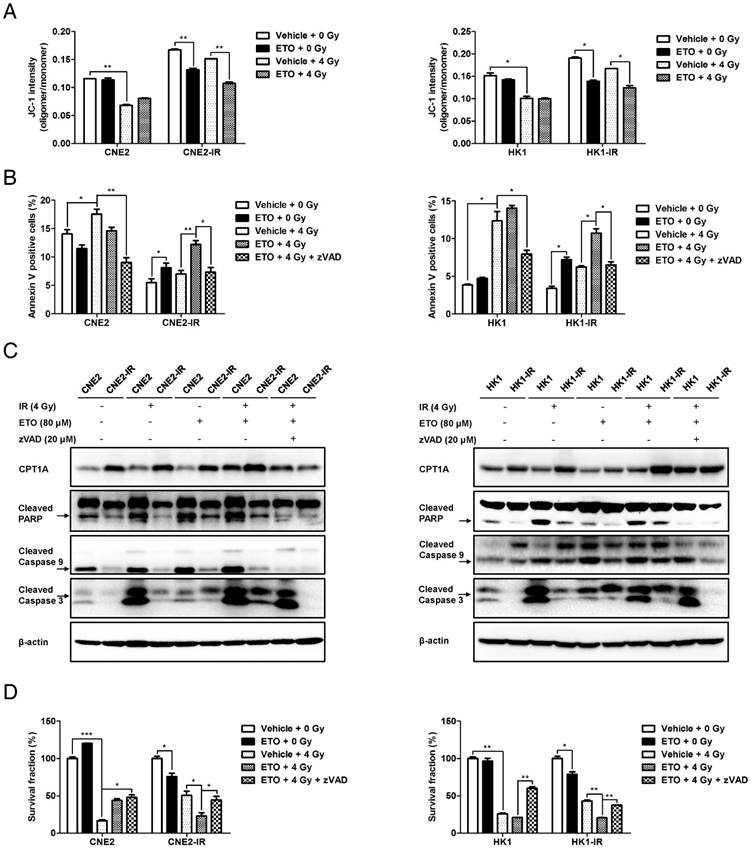
To investigate whether necroptosis and autophagy are involved in CPT1A inhibition-mediated cell death, we detected the protein levels of phosphorylated mixed lineage kinase domain-like (p-MLKL) and microtubule-associated protein 1 light chain 3 (LC3). Phosphorylation of MLKL is considered to be a hallmark of necroptosis and the conversion of LC3 I to LC3/II has been used has an indicator of autophagy [52]. TNFα plus Smac mimetic and z-VAD (T/S/Z) are classic inducers of necroptosis, and the human HT29 colon cancer cell line is one of the most sensitive and responsive cell lines to T/S/Z treatment [53]. Thus, we used the protein of HT29 cells treated with T/S/Z as a positive control of necroptosis. The results showed that none of the NPC cells following IR or ETO treatment underwent necroptosis (Figure S4A). Through the calculation of the LC3 II/I ratio, we found that ETO treatment suppressed autophagy in radiation-resistant cells (Figure S4A). A similar result was observed in CNE2-IR cells with CPT1A knockdown (Figure S4B). Autophagy is considered to be important for maintaining the functional pool of mitochondria for FAO, especially in stress conditions [54]. On the other hand, inhibition of FAO blocks lipid catabolism and might also have side effects on autophagy. However, the relationship between the two pathways and the role of autophagy in promoting radiation resistance need to be further explored.
To determine whether the anti-apoptotic effects mediated by CPT1A contribute to radiation resistance, zVAD was used in a colony formation assay combined with other treatments. Results showed that zVAD almost eliminates the inhibitory effects of ETO on cell survival in radiation-resistant cells (Figure 4D). A similar phenomenon was also observed in CNE2-IR cells with CPT1A knockdown (Figure S4C-E). These data demonstrate that inhibition of CPT1A by either pharmacological blockade or RNA interference could activate mitochondrial apoptosis, which re-sensitizes radiation-resistant NPC cells to radiation therapy.
CPT1A binds to Rab14 and promotes fatty acid trafficking in radiation-resistant NPC cells
We used a liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) approach to identify CPT1A-binding proteins. Thirty-four CPT1A-binding proteins are specific to CNE2-IR cells and 8 are Rab GTPases (Table S3). Based on the number of unique peptides and the protein score, we found that Rab7A and Rab14 were the top two of the 34 proteins. Immunoprecipitation analysis showed that CPT1A binds to Rab14, but not Rab7A, in NPC cells, and the binding of CPT1A-Rab14 is markedly increased in radiation-resistant cells (Figure 5A and Figure S5A). We also found that CPT1A binds to Rab14 in stable CPT1A-V5-overexpressing cells (Figure 5B). To explore whether radiation treatment could have an effect on the binding of CPT1A and Rab14, we performed immunoprecipitation analysis in response to a 4 Gy dose of radiation. The results showed that the binding of CPT1A and Rab14 is not influenced by radiation (Figure S5B). Nevertheless, we still treated cells with 4 Gy dose of radiation in the following study to create a stress condition, which was used previously. In addition to immunoprecipitation analysis, immunofluorescence assays showed that CPT1A co-localizes with Rab14 in radiation-resistant NPC cells (Figure S5C). To further confirm the interaction of the two proteins, we used an in situ proximity ligation assay (PLA). The assay generates a positive signal only when the distance between the two proteins is less than 40 nm and is highly suited for the direct detection of protein interactions [55]. CPT1A is reported to form a complex with voltage-dependent anion channel (VDAC) [56]. Thus, the detection of CPT1A and VDAC interaction served as a positive control and detection with only the CPT1A primary antibody served as a negative control. In this approach, radiation-resistant cells exhibited absolutely stronger positive fluorescence signals than radiation-responsive cells in response to radiation treatment (Figure 5C). These results confirm that CPT1A binds to Rab14 in NPC cells and the interaction is enhanced in radiation-resistant cells.
The binding of CPT1A to Rab14 is enhanced in radiation-resistant NPC cells, which might promote the contact of lipid droplet and mitochondria in these cells. (A) Immunoprecipitation analysis showing the binding of CPT1A and Rab14 in CNE2-IR and HK1-IR compared with parental cells. IgG served as a negative control. (B) Immunoprecipitation analysis showing the binding of CPT1A and Rab14 in CNE2 and HK1 cells stably expressing CPT1A-V5 or empty vector. IgG served as a negative control. (C) Proximity ligation assay indicating the interaction of CPT1A and Rab14 in CNE2-IR and HK1-IR compared with parental cells at 24 h after exposure to 4 Gy IR (red: PLA positive signal; blue: DAPI, scale bar = 25 μm). (D) Based on the prediction of Fd-DCA, the N terminal of CPT1A was found to bind with the identified Site 2 of Rab14 (left) and the predicted top 20 strongest coupled interacting residue pairs (interacting residue contacts) of these two proteins (orange stick bonds, right). (E) Immunoblot analysis showing translocation of CPT1A to mitochondria in CNE2 and CNE2-IR cells transfected with a Rab14 siRNA pool or negative siRNA at 24 h after exposure to 4 Gy IR. β-Actin was used as a control to confirm equal loading of protein and Hsp60 served as a control to confirm equal loading of mitochondrial fractions. (F) Confocal microscopy analysis of the co-localization of Rab14 and lipid droplets in CNE2-IR and HK1-IR cells and parental cells at 24 h after exposure to 4 Gy IR (red: Rab14; green: BODIPY 493/503; blue: DAPI; scale bar = 25 μm). (G) Confocal microscopy analysis of the co-localization of CPT1A, Rab14 and mitochondria in the groups indicated in (F) (red: Rab14; green: BODIPY 493/503; blue: DAPI; scale bar = 25 μm).
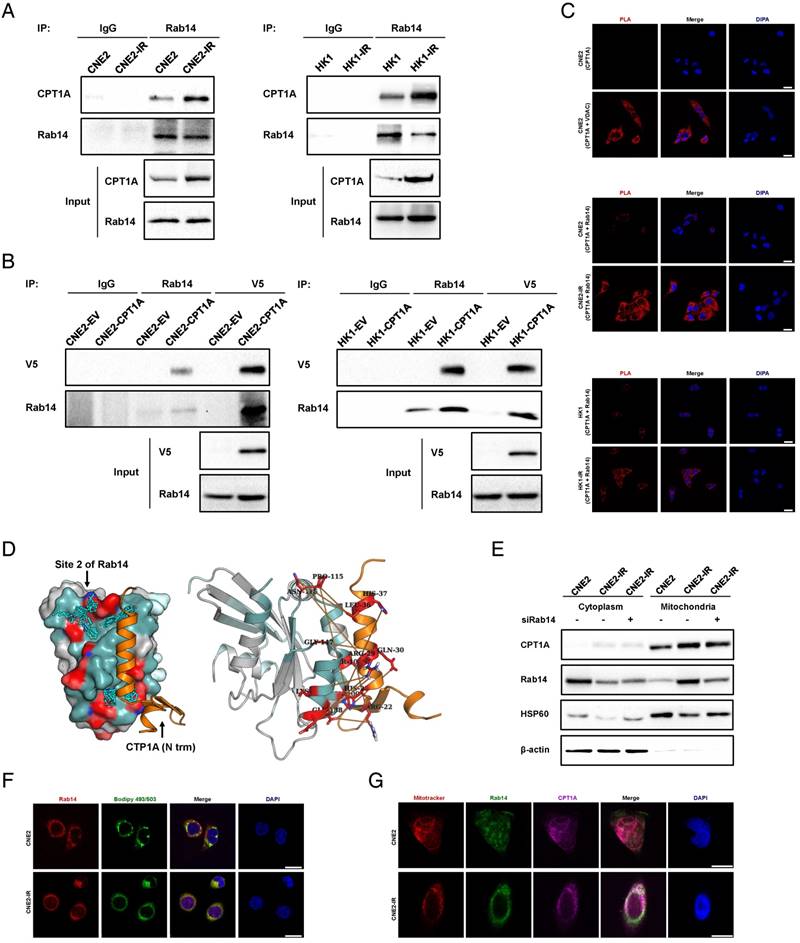
Next, we utilized a fragment docking and direct coupling combined computational protein-protein interaction prediction method, Fd-DCA, to predict the binding interface between CTP1A and RAB14 [57]. Two candidate sites, referred to as Site 1 and Site 2, were identified on the Rab14 surface (Figure S4D). According to the interfacial interaction score (computed based on the direct coupling analysis method), a strong coupling signal is observed between Site 2 and the alpha helix of the N terminal domain of CTP1A, indicating that the N terminal of CTP1A could bind to Site 2 of Rab14 (Figure 5D, Figure S4E-F and Table S4-5) [58]. Interestingly, we found that the predicted Site 1 is the binding site for Rab14 and Rab-coupling protein (RCP), which has been validated by a crystallized complex (PDB ID: 4d0g, Figure S4G) [59]. These in silico data provide evidence supporting the physical interaction between CPT1A and Rab14. Also, through the co-expression analysis of CPT1A versus Rab14 in NPC tissues from the Oncomine Database, we found that the mRNA level of Rab14 is positively correlated with the mRNA level of CPT1A (Figure S4H). For defining a crucial role of Rab14 in vesicle and protein transport, we examined the effects of Rab14 on CPT1A subcellular translocation in response to radiation [60]. CPT1A is mainly co-localized with mitochondria in radiation-resistant cells (Figure S4I). However, knockdown of Rab14 by siRNA does not alter the location pattern of CPT1A on mitochondria following radiation (Figure 5E). Rab GTPases are also reported to be essential for cell structures like lipid droplets and autophagosomes [61-63]. We found that Rab14 co-localizes with BODIPY 493/503-labled lipid droplets in NPC cells (Figure 5F). In addition, the co-localization of CPT1A, Rab14 and mitochondria is substantially increased in radiation-resistant NPC cells, which indicates that the binding of CPT1A and Rab14 might act as a 'bridge' between lipid droplets and mitochondria (Figure 5G).
Rab14 is essential to maintain the function of CPT1A in regulating fatty acid trafficking and radiation resistance
To investigate whether the CPT1A-Rab14 interaction plays a role in CPT1A-mediating radiation resistance, we disrupted the expression of Rab14 in CPT1A-overepressing cells. By using a Rab14 siRNA pool, the mRNA and protein levels of Rab14 are clearly attenuated in CPT1A- overexpressing and control cells (Figure S5A-B). Knockdown of Rab14 decreases PA-based OCR, induces apoptosis, and markedly suppresses radiation resistance in CPT1A-overexpressing cells compared with control cells following radiation treatment (Figure 6A and Figure S6C-D). The results suggest that Rab14 is essential to maintain the function of CPT1A in regulating FAO and radiation resistance.
Knockdown of Rab14 re-sensitizes CPT1A-overexpressing NPC cells to radiation therapy, suppresses fatty acid trafficking from lipid droplets into mitochondria and induces accumulation of lipid droplets in these cells. (A) Colony formation assay showing survival fractions of CNE2-EV, CNE2-CPT1A, HK1-EV and HK1-CPT1A cells transfected with a Rab14 siRNA pool or negative siRNA after exposure to 4 Gy IR. Survival fractions were calculated by comparing the colony number of each treatment group with untreated groups. Results are plotted as the mean survival fraction ± S.D. of 3 independent experiments (*p < 0.05, ***p < 0.001). (B) Confocal microscopy analysis of the morphologies and subcellular location of free fatty acids in CNE2-EV and CNE2-CPT1A cells that were chased for 0 or 12 h (red: Mito Tracker Red; green: BODIPY C16; blue: DAPI; scale bar = 25 μm). (C) Pearson's coefficient analysis representing relative cellular co-localization of free fatty acids overlapping with mitochondria in the groups indicated in (B). Images were analyzed using Image J software. Data are expressed as mean values ± S.D. of 6 independent cells analyzed by microscopy for each group (***p < 0.001). (D) Flow cytometry analysis of cellular neutral lipid content using the LipidTOX Red probe in cells indicated in (B) at 24 h after exposure to 4 Gy IR. Values represent LipidTOX Red intensity mean values ± S.D. of three independent experiments (*p < 0.05, **p < 0.01). (E) Intracellular ATP levels of the groups indicated in (B). Values represent ATP concentration mean values ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
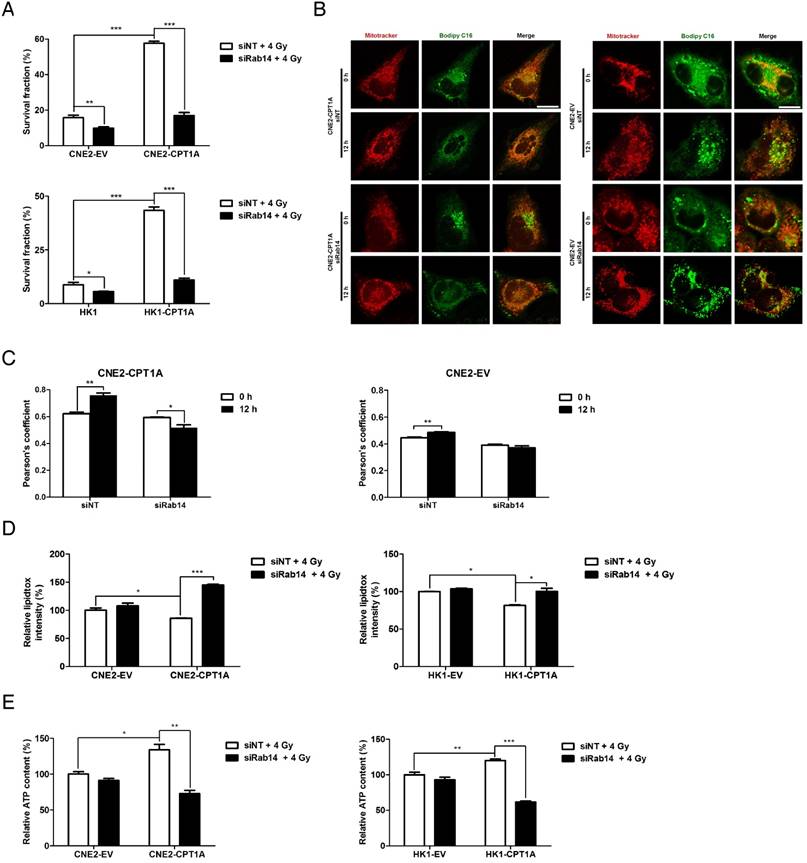
Based on the subcellular location of CPT1A and Rab14, the CPT1A-Rab14 interaction might provide contact between mitochondria and lipid droplets, which could facilitate fatty acid transport. To study fatty acid trafficking, we utilized BODIPY C16, a fatty acid analog bound with a BODIPY fluorophore, and a pulse-chase assay to track fatty acids in relation to mitochondria in response to radiation treatment. In this approach, all of the cells were treated with a 4 Gy dose of radiation and harvested at 24 h. The results showed that fatty acids are distributed homogeneously and mostly co-localize with mitochondria in CPT1A-overexpressing CNE2 cells after 12 h of chase; but, in control cells, fatty acids accumulate as lipid droplets, which seldom co-localize with mitochondria (Figure 6B-C). Knockdown of Rab14 decreases the translocation of fatty acids into mitochondria in CPT1A-overexpressing CNE2 cells. Similar results are observed in CPT1A-overexpressing HK1 cells (Figure S6E-F).
As a consequence of an impaired lipid flux after Rab14 knockdown, the content of lipid droplets is increased, whereas the ATP content is decreased in CPT1A-overexpressing cells following radiation (Figure 6D-E). In addition to CPT1A-overexpressing NPC cells, we also observed that knockdown of Rab14 suppresses cell survival in radiation-resistant NPC cells, which endogenously express high levels of CPT1A in response to radiation (Figure S7A-D). Overall, these results indicate that Rab14 is required to maintain the function of CPT1A in regulating fatty acid trafficking and radiation resistance.
Etomoxir disrupts the CPT1A-Rab14 interaction and fatty acid trafficking in radiation-resistant NPC cells
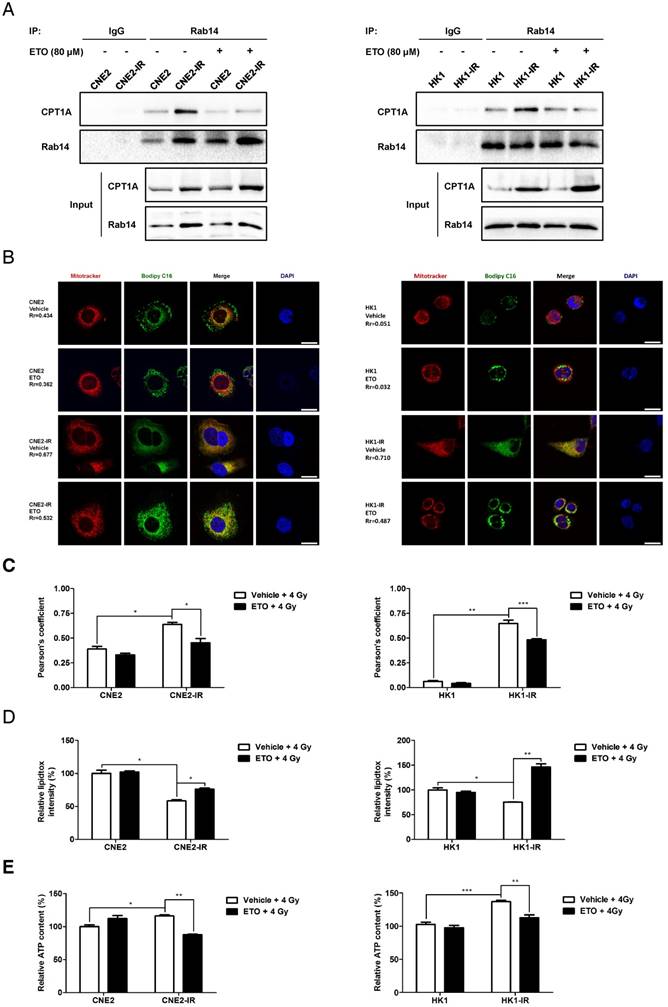
A previous study showed that ETO irreversibly binds to the active site on the C-terminal of the CPT1A protein and suppresses the enzymatic activity of CPT1A [64]. Surprisingly, in our study, we found that ETO disrupts the interaction of CPT1A and Rab14 in radiation-resistant NPC cells (Figure 7A). In the study of fatty acid trafficking, we found that fatty acids are homogenously distributed throughout the mitochondrial networks after a 4 Gy dose of radiation. However, ETO treatment blocks the translocation of fatty acids from lipid droplets into mitochondria in radiation-resistant cells (Figure 7B-C). Subsequently, lipid accumulation and ATP depletion are also observed in these cells upon ETO treatment with radiation treatment (Figure 7D-E). These results indicate that ETO might reduce the contact of lipid droplets and mitochondria by suppressing the CPT1A-Rab14 interaction, and it also decreases the transfer of fatty acids into the mitochondrial matrix by inhibiting CPT1A enzymatic activity following radiation therapy (Figure 9). In these ways, Etomoxir impairs fatty acid flux and augments radiation-induced energy starvation in radiation-resistant cells.
Etomoxir decreases the binding of CPT1A and Rab14, which attenuates fatty acid trafficking in radiation-resistant NPC cells. (A) Immunoprecipitation analysis showing the binding of CPT1A and Rab14 in CNE2-IR and HK1-IR compared with parental cells at 24 h after exposure to 4 Gy IR or Etomoxir (80 μM). IgG served as a negative control. (B) Confocal microscopy analysis of morphologies and subcellular location of free fatty acids in groups indicated in (A) (red: Mito Tracker Red; green: BODIPY C16; blue: DAPI; scale bar = 25 μm). (C) Pearson's coefficient analysis representing relative cellular co-localization of free fatty acids overlapping with mitochondria in groups indicated in (A). Images were analyzed using Image J software. Data are expressed as mean values ± S.D. of 12 independent cells analyzed by microscopy for each group (*p < 0.05, **p < 0.01, ***p < 0.001). (D) Flow cytometry analysis of cellular neutral lipid content using the LipidTOX Red probe in groups indicated in (A). Values represent LipidTOX Red intensity mean values ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01). (E) Intracellular ATP levels of groups indicated in (A). Values represent ATP concentration mean values ± S.D. of 3 independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001).
Furthermore, knockdown of CPT1A also induces lipid accumulation and ATP depletion in radiation-resistant NPC cells following radiation (Figure S8A-B). To explore whether the energy stress caused by CPT1A inhibition could be rescued by activation of other metabolic pathways, we supplemented 25 mM glucose or 5 mM glutamine to CPT1A knockdown cells and measured their ATP production at 24 h after exposure to radiation. The data show that glucose had no effect on ATP levels in response to radiation, but supplementation with glutamine dramatically enhanced ATP production in control cells, but only slightly increased the ATP levels in CPT1A knockdown cells (Figure S8C). The results indicate that glutamine could alleviate the energy crisis caused by CPT1A inhibition, but could not overcome it completely.
Etomoxir re-sensitizes NPC xenografts to radiation therapy
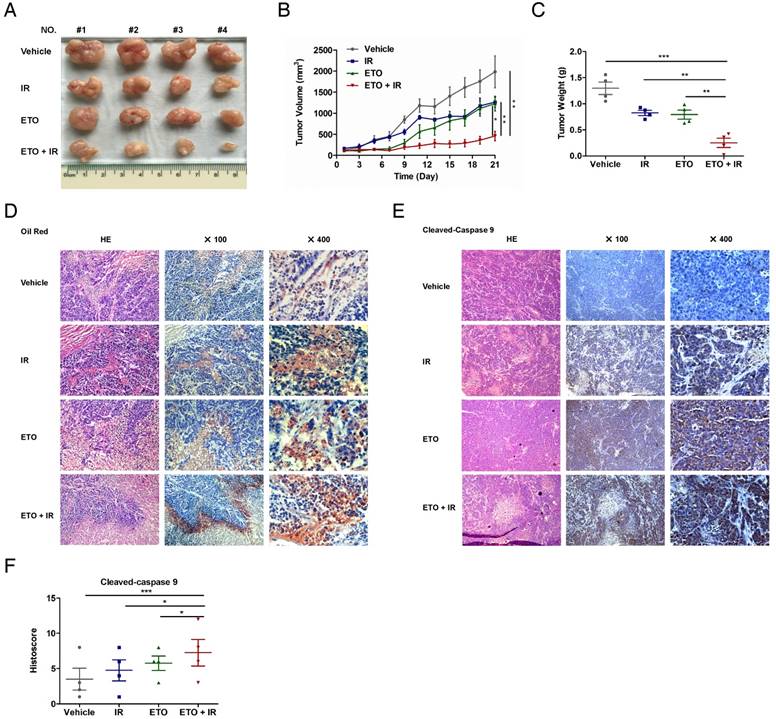
Based on the data obtained from in vitro models, we hypothesized that CPT1A confers radiation-resistance to NPC cells by preventing radiation-induced lipid accumulation and mitochondrial apoptosis. To confirm this hypothesis in vivo, we treated athymic nude mice bearing CNE2-IR xenografts with ETO, radiation (IR), or a combination of ETO and IR. CNE2 xenografts treated with radiation were also established as a radiation-responsive control. Consistent with the in vitro results, CNE2-IR xenografts were apparently more resistant to radiation than the CNE2 xenografts (Figure 8A-C and Figure S9A-C). Treatment with either radiation or ETO modestly decreased the tumor growth of CNE2-IR xenografts, whereas ETO combined with radiation substantially reduced tumor growth (Figure 8A-C and Figure 9D). These results indicate that ETO re-sensitizes CNE2-IR xenografts to radiation.
Etomoxir triggers intracellular lipid accumulation and apoptosis in vivo, and sensitizes tumors to radiation therapy. (A) Representative images of CNE2-IR xenografts from the indicated treatment groups. Vehicle: saline vehicle; IR: 2 Gy of irradiation restricted to tumors, twice a week, 2 weeks; ETO: Etomoxir, 40 mg/kg, every two days, 3 weeks; ETO + IR: Etomoxir-irradiation combination. (B) Tumor growth curves of CNE2-IR xenografts in groups indicated in (A). Tumor volume analysis is represented as mean values ± S.D. (n = 4, each group). Statistical significance of final tumor volumes was calculated using the two-tailed t test (*p < 0.05, **p < 0.01). (C) Tumor weight was measured at the end of the experiment. Results are shown as mean values ± S.D. (n = 4, each group; **p < 0.01, ***p < 0.001). (D) Representative Oil Red O staining of tumor xenografts from groups indicated in (A). (E) Representative cleaved caspase 9 staining of CNE2-IR xenografts in groups indicated in (A). (F) Column scatter of cleaved caspase 9 histoscores of CNE2-IR xenografts in groups indicated in (A). Results are shown as mean values ± S.D. (n = 4, each group; *p < 0.05, ***p < 0.001).
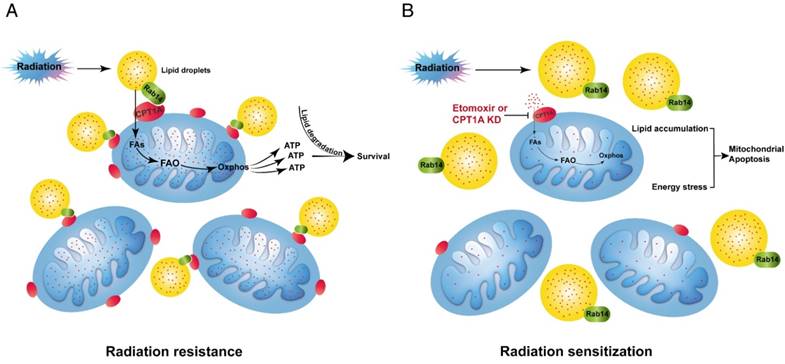
A schematic to illustrate CPT1A-mediated radiation resistance in NPC. (A) The enhanced CPT1A expression and the CPT1A-Rab14 interaction promote fatty acid trafficking between lipid droplets and mitochondria, which facilitates fatty acid utilization and maximizes ATP production, leading to resistance to radiation. (B) Blockage of CPT1A attenuates fatty acid trafficking and FAO, causes lipid accumulation and energy stress, which induces mitochondrial apoptosis and re-sensitizes NPC cells to radiation.
To further explore the association of lipid metabolism and radiation resistance, we stained lipid droplets in frozen tissue sections of CNE2-IR xenografts using Oil Red O. Most of the lipid droplets were distributed in tumor cells, and a few of them were scattered in mesenchymal tissue (Figure 8D). Surprisingly, the lipid droplets were abundant in tumor cells especially located on the edge of the typical cell death region, where the cells are found with homogeneous cytoplasm, pyknotic nuclei and lack of cell structures under hematoxylin and eosin staining. These histological changes combined with Oil Red O staining strongly suggest that lipid accumulation is associated with radiation-induced cell death. Moreover, ETO or radiation treatment increases the strength and positive rate of both Oil Red O and cleaved caspase 9, and the combination of ETO and radiation further enhances these alterations (Figure 8D-F). Based on these results, the accumulation of lipid droplets caused by CPT1A inhibition might contribute to mitochondrial apoptosis, which re-sensitizes CNE2-IR xenografts to radiation therapy (Figure 9).
Discussion
Tumor cells show metabolic flexibility in order to survive therapy [11, 65]. In recent years, metabolomics represents a promising approach to investigate the metabolic features of therapy-resistant tumor cells [66, 67]. Integrative metabolomics analysis was applied to discover the adaptive nutrients usage and specific metabolic pathway-addition in metformin-tolerant ovarian cancer and KRAS-ablation-resistant pancreatic cancer [68-70]. These metabolomics analyses provide fundamental insights into the metabolic preferences of therapy-resistant tumor cells and are helpful to identify new drug targets to improve therapeutic outcomes.
In this study, we performed metabolomics and identified a robust lipid turnover and FAO signature in radiation-resistant NPC cells, which suggests that lipid reprogramming may play a vital role in mediating radiation resistance in NPC. Previous studies focusing on lipid metabolism in NPC reported a unique fatty acid synthesis (FAS) profile in NPC [71, 72]. Compared with nasopharyngeal epithelium, NPC tissue exhibited strong accumulation of lipid droplets [72]. Moreover, adipocyte triglyceride lipase (ATGL) was down-regulated in NPC cell lines relative to nasopharyngeal epithelial cell lines [72]. However, an active lipolysis and high expression of monoglyceride lipase (MAGL) was detected in highly metastatic NPC cells relative to a low-metastatic population [73]. We also found that radiation-resistant NPC cells have an active fatty acid trafficking and utilization signature compared with radiation-responsive cells. These lipid metabolic characteristics in highly metastatic NPC cells and radiation-resistant NPC cells indicate that the metabolic shift from FAS to lipolysis may provide a survival advantage for NPC cells to overcome anoikis or radiation-induced stress.
Based on the metabolomics data, CPT1A, the rate-limiting enzyme of FAO, is consistently up-regulated in radiation-resistant NPC cells, and high expression of CPT1A is significantly associated with poor overall survival of NPC patients following radiation therapy. Moreover, inhibition of CPT1A re-sensitizes NPC cells to radiation therapy in vitro and in vivo. The evidence suggests that the expression of CPT1A could be a prognostic indicator of NPC, and targeting FAO in CPT1A high-expression NPC patients might improve the therapeutic efficacy of radiotherapy.
Previous studies showed that CPT1A could form a complex with long chain acyl-CoA synthetase (ACSL) and voltage-dependent anion channel (VDAC) on mitochondrial outer membranes to benefit fatty acid transportation through the mitochondrial membrane [56]. Acetyl-CoA carboxylase α (ACC), the first enzyme involved in FAS, also interacts with CPT1A in hepatocellular carcinoma [74]. Glucose starvation dissociated CPT1A from the complex and increased the translocation of CPT1A onto mitochondria, which suggests that a transformation from FAS to FAO is needed to overcome nutrient depletion. According to our LC-MS/MS analysis, we did not find the binding of ACC or ACSL to CPT1A in NPC cells, but we found that VDAC and, intriguingly, several Rab GTPases potentially bind to CPT1A. We observed an increased binding of CPT1A-Rab14 in radiation-resistant NPC cells. Additionally, the mRNA level of Rab14 was positively correlated with the mRNA level of CPT1A in NPC tumor tissues from the Oncomine Database. Rab GTPases belong to the RAS oncogene family, which regulates vesicle trafficking and receptor cycling [75, 76]. Rab14 is reported to be directly targeted by microRNA-451 and microRNA-338-3p in human non-small-cell lung cancer [77, 78]. Recently, Rab14 was found to be up-regulated in gastric cancer cells and promotes cell proliferation [79]. However, the mechanism of Rab14 involvement in cancer is still unclear.
In our study, we found that Rab14 does not affect the translocation of CPT1A onto mitochondria in NPC cells. However, it could locate on lipid droplets in NPC cells, consistent with three lipid droplet-specific proteomics screenings in other cells [63]. Recently, lipid droplets have been recognized as active organelles [80]. The close physiological association between lipid droplets and mitochondria has been confirmed in response to nutrient depletion [81, 82]. In our study, the co-localization of CPT1A, Rab14 and mitochondria was remarkably increased in radiation-resistant NPC cells. Knockdown of Rab14 re-sensitized CPT1A-overexpressing NPC cells to radiation, and decreased fatty acid transfer from lipid droplets to mitochondria in these cells. These results indicate that the binding of CPT1A and Rab14 might act as a molecular bridge between lipid droplets and mitochondria and promotes fatty acid trafficking.
In addition to the function of CPT1A in ATP and NADPH production, a new role for CPT1A in epigenetics has been identified [29, 31, 32]. The mitochondria-generated acetyl coenzyme A could be exported to the cytoplasm through the citrate transporter and citrate lyase. The ratio of cytosolic acetyl coenzyme A is essential for histone acetylation by p300 [83]. Recently, CPT1A-dependent acetyl coenzyme A production was found to be essential to promote acetylation of histones at lymphangiogenic genes [84, 85]. In another study, CPT1A replenished carbons of the TCA cycle that were incorporated into nucleotide precursors and substantially promoted nucleotide synthesis in endothelial cells [86]. Intriguingly, a product of CPT1A transcript variant 2, was found to be expressed in the nuclei of breast cancer cells, which interacted with histone deacetylase 1 (HDAC1), leading to epigenetic regulation of cancer-related gene expression [87]. The function of CPT1A in epigenetics and nucleotide synthesis might also contribute to tumor therapeutic resistance.
Due to the critical role of CPT1 in diseases, drugs specifically targeting CPT1 have begun to receive the attention of researchers. The most investigated small-molecule inhibitor of CPT1 enzyme is Etomoxir, which has been used in the clinical treatment of type 2 diabetes and heart failure [49, 88-90]. In our study, Etomoxir efficiently re-sensitized NPC cells to radiation therapy in vitro and in vivo. Other studies also reported that Etomoxir exerted anticancer effects against prostate cancer, colon cancer, metastatic triple-negative breast cancer and leukemia in vivo [34, 91-93].
We unexpectedly found that Etomoxir has inhibitory effects on the CPT1A-Rab14 interaction, but the mechanism is unclear. A previous study showed that Etomoxir irreversibly binds to the active site on the C-terminal of the CPT1A protein [64]. Our computational protein-protein interaction prediction only suggested a physical interaction between the N terminal of CPT1A and Rab14. Thus, whether Etomoxir directly targets the binding sites of CPT1A and Rab14 or changes the conformation of the CPT1A protein and subsequently suppresses the CPT1A-Rab14 interaction is not clear. In the future, further experiments should be performed to delineate the precise mechanism of the CPT1A-Rab14 interaction.
Overall, through a metabolomics approach, we found that FAO is active in radiation-resistant NPC cells. Additionally, the rate-limiting enzyme of FAO, CPT1A, is highly expressed in radiation-resistant NPC cells and corresponds with poor overall survival of NPC patients following radiation therapy. Importantly, we identified a lipid-associated protein, Rab14, as a novel CPT1A binding protein. The CPT1A-Rab14 interaction facilitates fatty acid transfer from lipid droplets to mitochondria, which promotes fatty acid utilization and maximizes ATP production. Knockdown of Rab14 attenuates CPT1A-mediated fatty acid trafficking and radiation resistance. Inhibition of CPT1A re-sensitizes radiation-resistant NPC cells to radiation therapy through the induction of lipid accumulation and mitochondrial apoptosis both in vitro and in vivo. Collectively, our data demonstrate the utility of metabolomics approaches for addressing cancer therapy resistance, and suggest that targeting CPT1A might be a beneficial regimen to improve the therapeutic effects of radiotherapy in NPC patients. Additionally, the CPT1A-Rab14 interaction could be another attractive interface for the discovery of novel CPT1A inhibitors.
Abbreviations
ACC: acetyl-CoA carboxylase α; ACSL: long chain acyl-CoA synthetase; ATGL: adipocyte triglyceride lipase; ATP: adenosine triphosphate; BSA: bovine serum albumin; CPT1A: carnitine palmitoyl transferase 1A; EBER: Epstein-Barr Virus-encoded RNA; ETO: ethyl 2-[6-(4-chlorophenoxy)hexyl] oxirane-2-carboxylate; FAO: fatty acid oxidation; FAS: fatty acid synthesis; HADA1: histone deacetylase 1; IC50: concentration of compound causing 50% inhibition; LC-MS/MS: liquid chromatography coupled with tandem mass spectrometry; MAGL: monoglyceride lipase; MMP: mitochondrial membrane potential; NADPH: nicotinamide adenine dinucleotide; NPC: nasopharyngeal carcinoma; Oxphos: oxidative phosphorylation; PA: palmitate; PLA: proximity ligation assay; RCP: Rab-coupling protein; shRNA: short hairpin RNA; siRNA: small-interfering RNA; TCA: tricarboxylic acid; VDAC: voltage-dependent anion channel; zVAD: z-Val-Ala-Asp-fmk.
Acknowledgements
The study was supported by National Science Foundation of China, No. 81573014; National Science Foundation of China, No. 81402250; Key Projects of National Key Research and Development Program in China (2016YFC0902000). Work at the Center for Theoretical Biological Physics was sponsored by the National Science Foundation (Grants PHY-1427654) and the Cancer Prevention and Research Institute of Texas (CPRIT-grant R1110). F.B. was supported by Welch Foundation Grant C-1792.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chua ML, Wee JT, Hui EP. et al. Nasopharyngeal carcinoma. Lancet. 2016;387(10022):1012-24
2. Mao YP, Xie FY, Liu LZ. et al. Re-evaluation of 6th edition of AJCC staging system for nasopharyngeal carcinoma and proposed improvement based on magnetic resonance imaging. Int J Radiat Oncol Biol Phys. 2009;73(5):1326-34
3. Rottey S, Madani I, Deron P. et al. Modern treatment for nasopharyngeal carcinoma: current status and prospects. Curr Opin Oncol. 2011;23(3):254-8
4. Lu J, Tang M, Li H. et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016;380(1):191-200
5. Cooper JS, Ang KK. Concomitant chemotherapy and radiation therapy certainly improves local control. Int J Radiat Oncol Biol Phys. 2005;61(1):7-9
6. Liao K, Xia B, Zhuang QY. et al. Parthenolide inhibits cancer stem-like side population of nasopharyngeal carcinoma cells via suppression of the NF-kappaB/COX-2 pathway. Theranostics. 2015;5(3):302-21
7. Ma J, Mai HQ, Hong MH. et al. Results of a prospective randomized trial comparing neoadjuvant chemotherapy plus radiotherapy with radiotherapy alone in patients with locoregionally advanced nasopharyngeal carcinoma. J Clin Oncol. 2001;19(5):1350-7
8. Hareyama M, Sakata K, Shirato H. et al. A prospective, randomized trial comparing neoadjuvant chemotherapy with radiotherapy alone in patients with advanced nasopharyngeal carcinoma. Cancer. 2002;94(8):2217-23
9. Chua DT, Sham JS, Choy D. et al. Preliminary report of the Asian-Oceanian Clinical Oncology Association randomized trial comparing cisplatin and epirubicin followed by radiotherapy versus radiotherapy alone in the treatment of patients with locoregionally advanced nasopharyngeal carcinoma. Asian-Oceanian Clinical Oncology Association Nasopharynx Cancer Study Group. Cancer. 1998;83(11):2270-83
10. Yi L, Song C, Hu Z. et al. A metabolic discrimination model for nasopharyngeal carcinoma and its potential role in the therapeutic evaluation of radiotherapy. Metabolomics. 2014;10(4):697-708
11. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351-9
12. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27-47
13. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364-73
14. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-14
15. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-33
16. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297-308
17. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678-84
18. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9-15
19. Currie E, Schulze A, Zechner R. et al. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153-61
20. Xiao L, Hu ZY, Dong X. et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33(37):4568-78
21. Chen X, Wei S, Ma Y. et al. Quantitative proteomics analysis identifies mitochondria as therapeutic targets of multidrug-resistance in ovarian cancer. Theranostics. 2014;4(12):1164-75
22. Kastaniotis AJ, Autio KJ, Keratar JM. et al. Mitochondrial fatty acid synthesis, fatty acids and mitochondrial physiology. Biochim Biophys Acta. 2017;1862(1):39-48
23. Vishwanath VA. Fatty Acid Beta-Oxidation Disorders: A Brief Review. Ann Neurosci. 2016;23(1):51-5
24. Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol. 2014;171(8):2080-90
25. Gregersen N, Olsen RK. Disease mechanisms and protein structures in fatty acid oxidation defects. J Inherit Metab Dis. 2010;33(5):547-53
26. Fukushima A, Lopaschuk GD. Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim Biophys Acta. 2016;1862(12):2211-20
27. Gariani K, Ryu D, Menzies KJ. et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J Hepatol. 2017;66(1):132-41
28. Choi J, Ravipati A, Nimmagadda V. et al. Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion. 2014;18:41-8
29. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227-32
30. Zechner R, Zimmermann R, Eichmann TO. et al. FAT SIGNALS-lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15(3):279-91
31. Zaugg K, Yao Y, Reilly PT. et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25(10):1041-51
32. Pike LS, Smift AL, Croteau NJ. et al. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta. 2011;1807(6):726-34
33. Ye H, Adane B, Khan N. et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell. 2016;19(1):23-37
34. Camarda R, Zhou AY, Kohnz RA. et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427-32
35. Caro P, Kishan AU, Norberg E. et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22(4):547-60
36. Luo X, Cheng C, Tan Z. et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76
37. Schreurs M, Kuipers F, van der Leij FR. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Rev. 2010;11(5):380-8
38. Shao H, Mohamed EM, Xu GG. et al. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget. 2016;7(4):3832-46
39. Li G, Liu Y, Su Z. et al. MicroRNA-324-3p regulates nasopharyngeal carcinoma radioresistance by directly targeting WNT2B. Eur J Cancer. 2013;49(11):2596-607
40. Li G, Qiu Y, Su Z. et al. Genome-wide analyses of radioresistance-associated miRNA expression profile in nasopharyngeal carcinoma using next generation deep sequencing. PLoS One. 2013;8(12e):84486
41. Qu JQ, Yi HM, Ye X. et al. MiRNA-203 Reduces Nasopharyngeal Carcinoma Radioresistance by Targeting IL8/AKT Signaling. Mol Cancer Ther. 2015;14(11):2653-64
42. Zhao L, Tang M, Hu Z. et al. miR-504 mediated down-regulation of nuclear respiratory factor 1 leads to radio-resistance in nasopharyngeal carcinoma. Oncotarget. 2015;6(18):15995-6018
43. Reitman ZJ, Jin G, Karoly ED. et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108(8):3270-5
44. Bieber LL, Abraham T, Helmrath T. A rapid spectrophotometric assay for carnitine palmitoyltransferase. Anal Biochem. 1972;50(2):509-18
45. Karlic H, Lohninger S, Koeck T. et al. Dietary l-carnitine stimulates carnitine acyltransferases in the liver of aged rats. J Histochem Cytochem. 2002;50(2):205-12
46. Linher-Melville K, Zantinge S, Sanli T. et al. Establishing a relationship between prolactin and altered fatty acid beta-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer. 2011;11:56
47. Hu J, Deng X, Bian X. et al. The expression of functional chemokine receptor CXCR4 is associated with the metastatic potential of human nasopharyngeal carcinoma. Clin Cancer Res. 2005;11(13):4658-65
48. Lopaschuk GD, Wall SR, Olley PM. et al. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63(6):1036-43
49. Holubarsch CJ, Rohrbach M, Karrasch M. et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond). 2007;113(4):205-12
50. Airoldi M, Garzaro M, Gabriele A. et al. Induction chemotherapy with cisplatin and epirubicin followed by radiotherapy and concurrent cisplatin in locally advanced nasopharyngeal carcinoma observed in a nonendemic population. J Clin Oncol. 2009;27(15_suppl):e17040
51. Chua DT, Ma J, Sham JS. et al. Long-term survival after cisplatin-based induction chemotherapy and radiotherapy for nasopharyngeal carcinoma: a pooled data analysis of two phase III trials. J Clin Oncol. 2005;23(6):1118-24
52. Ma XH, Piao SF, Dey S. et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124(3):1406-17
53. Cai Z, Zhang A, Choksi S. et al. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res. 2016;26(8):886-900
54. Guo JY, Karsli-Uzunbas G, Mathew R. et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447-61
55. Soderberg O, Gullberg M, Jarvius M. et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995-1000
56. Lee K, Kerner J, Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem. 2011;286(29):25655-62
57. Bai F, Morcos F, Cheng RR. et al. Elucidating the druggable interface of protein-protein interactions using fragment docking and coevolutionary analysis. Proc Natl Acad Sci U S A. 2016;113(50E):8051-E8
58. Morcos F, Pagnani A, Lunt B. et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc Natl Acad Sci U S A. 2011;108(49E):1293-301
59. Lall P, Lindsay AJ, Hanscom S. et al. Structure-Function Analyses of the Interactions between Rab11 and Rab14 Small GTPases with Their Shared Effector Rab Coupling Protein (RCP). J Biol Chem. 2015;290(30):18817-32
60. Zhen Y, Stenmark H. Cellular functions of Rab GTPases at a glance. J Cell Sci. 2015;128(17):3171-6
61. Bartz R, Zehmer JK, Zhu M. et al. Dynamic activity of lipid droplets: protein phosphorylation and GTP-mediated protein translocation. J Proteome Res. 2007;6(8):3256-65
62. Martin S, Driessen K, Nixon SJ. et al. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J Biol Chem. 2005;280(51):42325-35
63. Kiss RS, Nilsson T. Rab proteins implicated in lipid storage and mobilization. J Biomed Res. 2014;28(3):169-77
64. Rufer AC, Thoma R, Hennig M. Structural insight into function and regulation of carnitine palmitoyltransferase. Cell Mol Life Sci. 2009;66(15):2489-501
65. Tan Z, Luo X, Xiao L. et al. The Role of PGC1alpha in Cancer Metabolism and its Therapeutic Implications. Mol Cancer Ther. 2016;15(5):774-82
66. Puhka M, Takatalo M, Nordberg ME. et al. Metabolomic Profiling of Extracellular Vesicles and Alternative Normalization Methods Reveal Enriched Metabolites and Strategies to Study Prostate Cancer-Related Changes. Theranostics. 2017;7(16):3824-41
67. Larkin JR, Dickens AM, Claridge TD. et al. Early Diagnosis of Brain Metastases Using a Biofluids-Metabolomics Approach in Mice. Theranostics. 2016;6(12):2161-9
68. Liu X, Romero IL, Litchfield LM. et al. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016;24(5):728-39
69. Elgogary A, Xu Q, Poore B. et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A. 2016;113(36E):5328-36
70. Viale A, Pettazzoni P, Lyssiotis CA. et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628-32
71. Kao YC, Lee SW, Lin LC. et al. Fatty acid synthase overexpression confers an independent prognosticator and associates with radiation resistance in nasopharyngeal carcinoma. Tumour Biol. 2013;34(2):759-68
72. Zhou X, Wei J, Chen F. et al. Epigenetic downregulation of the ISG15-conjugating enzyme UbcH8 impairs lipolysis and correlates with poor prognosis in nasopharyngeal carcinoma. Oncotarget. 2015;6(38):41077-91
73. Hu WR, Lian YF, Peng LX. et al. Monoacylglycerol lipase promotes metastases in nasopharyngeal carcinoma. Int J Clin Exp Pathol. 2014;7(7):3704-13
74. Wang MD, Wu H, Fu GB. et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology. 2016;63(4):1272-86
75. Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2(2):107-17
76. Zhang J, Zhang X, Liu G. et al. Intracellular Trafficking Network of Protein Nanocapsules: Endocytosis, Exocytosis and Autophagy. Theranostics. 2016;6(12):2099-113
77. Wang R, Wang ZX, Yang JS. et al. MicroRNA-451 functions as a tumor suppressor in human non-small cell lung cancer by targeting ras-related protein 14 (RAB14). Oncogene. 2011;30(23):2644-58
78. Sun J, Feng X, Gao S. et al. microRNA-338-3p functions as a tumor suppressor in human nonsmallcell lung carcinoma and targets Ras-related protein 14. Mol Med Rep. 2015;11(2):1400-6
79. Guo B, Wang W, Zhao Z. et al. Rab14 Act as Oncogene and Induce Proliferation of Gastric Cancer Cells via AKT Signaling Pathway. PLoS One. 2017;12(1e):0170620
80. Welte MA. Expanding roles for lipid droplets. Curr Biol. 2015;25(11R):470-81
81. Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32(6):678-92
82. Herms A, Bosch M, Reddy BJ. et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun. 2015;6:7176
83. Lee JV, Carrer A, Shah S. et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20(2):306-19
84. Wong BW, Wang X, Zecchin A. et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature. 2017;542(7639):49-54
85. Hosios AM, Vander Heiden MG. Endothelial Cells Get beta-ox-ed In to Support Lymphangiogenesis. Dev Cell. 2017;40(2):118-9
86. Schoors S, Bruning U, Missiaen R. et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520(7546):192-7
87. Pucci S, Zonetti MJ, Fisco T. et al. Carnitine palmitoyl transferase-1A (CPT1A): a new tumor specific target in human breast cancer. Oncotarget. 2016;7(15):19982-96
88. Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci (Lond). 2000;99(1):27-35
89. Selby PL, Sherratt HS. Substituted 2-oxiranecarboxylic acids: a new group of candidate hypoglycaemic drugs. Trends Pharmacol Sci. 1989;10(12):495-500
90. Hernlund E, Ihrlund LS, Khan O. et al. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int J Cancer. 2008;123(2):476-83
91. Schlaepfer IR, Rider L, Rodrigues LU. et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther. 2014;13(10):2361-71
92. Samudio I, Harmancey R, Fiegl M. et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120(1):142-56
93. Park JH, Vithayathil S, Kumar S. et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016;14(9):2154-65
Author contact
![]() Corresponding authors: Prof. Ya Cao or Pro Xiangjian Luo, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China. Telephone: +86-731-84805448; E-mail: ycao98sina.com or luocsucom.
Corresponding authors: Prof. Ya Cao or Pro Xiangjian Luo, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China. Telephone: +86-731-84805448; E-mail: ycao98sina.com or luocsucom.