Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2019; 9(3):646-660. doi:10.7150/thno.30276 This issue Cite
Research Paper
Nuclear Entry of CRTC1 as Druggable Target of Acquired Pigmentary Disorder
Cheong-Yong Yun1,*, Seung Deok Hong1,*, Young Hee Lee2, Jiyeon Lee1, Da-Eun Jung1, Ga Hyun Kim1, Song-Hee Kim1, Jae-Kyung Jung1, Ki Ho Kim3, Heesoon Lee1, Jin Tae Hong1, Sang-Bae Han1, Youngsoo Kim1 ![]()
1. College of Pharmacy, Chungbuk National University, Cheongju 28160, Korea
2. Samjin Pharmaceutical Company, Seoul 04054, Korea
3. Kihobio Company, Cheongju 28160, Korea
* C.-Y.Y. and S.D.H. equally contributed to this study.
Received 2018-9-28; Accepted 2018-12-21; Published 2019-1-21
Abstract

Rationale: SOX10 (SRY-related HMG-box 10) and MITF-M (microphthalmia-associated transcription factor M) restrict the expression of melanogenic genes, such as TYR (tyrosinase), in melanocytes. DACE (diacetylcaffeic acid cyclohexyl ester) inhibits melanin production in α-MSH (α-melanocyte stimulating hormone)-activated B16-F0 melanoma cells. In this study, we evaluated the antimelanogenic activity of DACE in vivo and elucidated the molecular basis of its action.
Methods: We employed melanocyte cultures and hyperpigmented skin samples for pigmentation assays, and applied chromatin immunoprecipitation, immunoblotting, RT-PCR or siRNA-based knockdown for mechanistic analyses.
Results: Topical treatment with DACE mitigated UV-B-induced hyperpigmentation in the skin with attenuated expression of MITF-M and TYR. DACE also inhibited melanin production in α-MSH- or ET-1 (endothelin 1)-activated melanocyte cultures. As a mechanism, DACE blocked the nuclear import of CRTC1 (CREB-regulated co-activator 1) in melanocytes. DACE resultantly inhibited SOX10 induction, and suppressed the transcriptional abilities of CREB/CRTC1 heterodimer and SOX10 at MITF-M promoter, thereby ameliorating facultative melanogenesis. Furthermore, this study unveiled new issues in melanocyte biology that i) KPNA1 (Impα5) escorted CRTC1 as a cargo across the nuclear envelope, ii) SOX10 was inducible in the melanogenic process, and iii) CRTC1 could direct SOX10 induction at the transcription level.
Conclusion: We propose the targeting of CRTC1 as a unique strategy in the treatment of acquired pigmentary disorders.
Keywords: diacetylcaffeic acid cyclohexyl ester, CRTC1, SOX10, MITF-M, facultative melanogenesis
Introduction
The cAMP-responsive element (CRE)-binding protein (CREB), in concert with three isoforms of CRTCs (CRTC1-3), integrates cellular signals of cAMP and Ca2+ into transcriptional responses [1]. Following phosphorylation at Ser-133, CREB recruits CREB-binding protein (CBP) and p300 that acetylate nucleosomal histones in epigenetically active chromatin remodeling [2]. CRTCs do not directly bind to DNA [1]. They interact with the basic Leu zipper (bZIP) domain of CREB and co-activate the transcriptional ability of CREB, recruiting TAFII130 component of basal transcription factor TFIID followed by RNA polymerase II complex onto target promoters [1, 3]. Deregulation of CRTCs has been implicated in age-related disease risk [4].
In the current study, we propose the targeting of CRTC1 as a unique strategy in the treatment of acquired skin hyperpigmentation. The melanogenic process synthesizes melanin pigments, black-brown eumelanin and yellow-reddish pheomelanin, in melanosomes of melanocytes at the epidermal/ dermal border, and transfers pigmented melanosomes to keratinocytes in the overlaying epidermis, which determines the skin color, and shields the nuclei of skin cells from solar UV radiation [5, 6, 7]. An imbalance in this process results in acquired pigmentary disorder, and often in melanoma [5, 7, 8]. MITF-M plays important roles in the expression of melanogenesis enzymes such as TYR, TYR-related protein 1 (TYRP1) and dopachrome tautomerase (DCT), and also in the development and differentiation of melanocytes [9, 10]. TYR catalyzes a rate-liming step in the biosynthetic pathways of eumelanin and pheomelanin. TYRP1 and DCT are indispensable in the production of eumelanin, but not in that of pheomelanin [11]. However, MITF-M is not a druggable target in the melanocyte homeostasis, particularly in melanoma therapy [12]. MITF-M-targeted approaches are based on the modulation of its upstream regulatory pathways [12, 13].
Hormonal activation of G protein-coupled receptors (GPCRs), such as melanocortin 1 receptor (MC1R) and endothelin receptor B (ENDRB), results in heavily pigmented melanosomes in melanocytes [14]. When MC1R is bound by α-MSH, the Gαs protein stimulates adenylate cyclase activity and activates the pathways downstream to cAMP-protein kinase A (PKA) [14, 15]. α-MSH increases the intracellular Ca2+ levels in a cholera toxin-sensitive, but inositol 1,4,5-triphosphate (IP3)-independent manner [16]. In turn, PKA activates CREB activity via phosphorylation at Ser-133, and inhibits salt-inducible kinase (SIK) activity via phosphorylation at Ser-587 [17, 18]. SIK phosphorylates CRTC1, but the Ca2+-dependent activation of protein phosphatase 2B (PP2B) removes phosphate groups from CRTCs [1, 19]. The unphosphorylated (dephospho)-CRTC1 shuttles into the nucleus, while phosphorylated (p)-CRTC1 is sequestered in the cytoplasm [19, 20]. The p-CREB coupled with dephospho-CRTC1 lead to expression of target genes, such as MITF-M, that contain the CRE motif in their promoter region [1]. However, the influence of CREB or CRTC1 on the expression of SOX10 remains unknown, despite the central role of SOX10 in melanocyte-specific transcriptional activation of MITF-M from cAMP- and Ca2+-mediated signaling pathways that are ubiquitous in almost all cells [21, 22].
When ENDRB is bound by ET-1, the Gαq protein stimulates the activity of phospholipase Cβ (PLCβ), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into IP3 and diacylglycerol (DAG) [14]. This leads to increase intracellular Ca2+ levels and to activate PKC activity [14, 23]. In turn, PKC phosphorylates adenylate cyclase that links to activation of the cAMP-PKA pathway, and the rapidly accelerated fibrosarcoma (Raf) that links to downstream pathway of mitogen-activated protein kinase kinase (MEK)- extracellular signal-regulated kinase (ERK)-p90 ribosomal S6 kinase (RSK) [24]. The Raf-MEK-ERK-RSK pathway enhances the transcriptional activity of CREB or MITF-M, and protects SOX10 from degradation via specific phosphorylation [24, 25].
Diacetylcaffeic acid cyclohexyl ester (DACE, Figure 1A) inhibits melanin production in α-MSH-activated B16-F0 cells and its efficacy is decreased when cyclohexyl ester moiety is replaced by amide groups with a phenyl, piperazine or pyrazole ring [26]. In the current study, we evaluated the antimelanogenic activity of DACE in vivo and elucidated its mechanism of action. DACE mitigated UV-B-induced hyperpigmentation in the skin by blocking the nuclear import of CRTC1 in melanocytes. DACE resultantly inhibited SOX10 induction and suppressed the transcriptional abilities of CREB/CRTC1 heterodimer and SOX10 at the MITF-M promoter, thus preventing acquired hyperpigmentation.
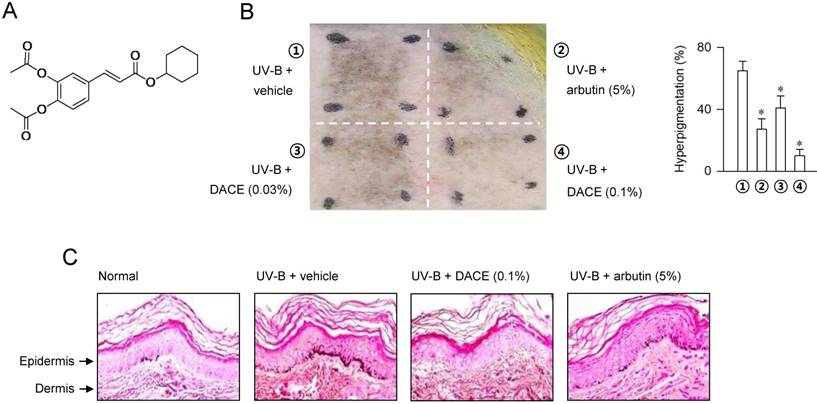
Effect of DACE on skin hyperpigmentation. (A) Chemical structure of DACE. Dorsal skins of guinea pigs were exposed to UV-B radiation (350 mJ/cm2) and treated topically with DACE according to the protocol in Figure S1. (B) A representative photograph of pigmented skins and its relative pigmentation change (% of treated area) are presented. Data are mean ± SEM (n = 5). *P < 0.05 vs. UV-B plus vehicle alone. (C) Skin tissues were sectioned and then reacted with Fontana-Masson silver nitrate to stain melanin granules.
Results
DACE mitigated UV-B-induced hyperpigmentation in the skin
In order to understand the antimelanogenic activity in vivo, dorsal skins of guinea pigs were exposed to UV-B (350 mJ/cm2) and treated topically with DACE (0.03-0.1%) or arbutin (5%, a dose recommended by Korean Ministry of Food and Drug Safety, KFDA) according to the protocol in Figure S1. Topical treatment with DACE attenuated the visual hyperpigmentation in the absence of corrosion, as did arbutin (Figure 1B). Arbutin, a KFDA-approved skin whitener that was employed as a positive control in this study, inhibits catalytic activity of TYR in a competitive mechanism with respect to substrate [27]. Furthermore, skin tissues were sectioned and then stained with Fontana-Masson silver nitrate (Figure 1C). DACE consistently decreased UV-B-induced melanin granules in the skin, more evident in the basal layer of epidermis where generates the pigmented melanosomes in melanocytes (Figure 1C). Thus, DACE could mitigate the acquired hyperpigmentation in UV-B-exposed skins.
DACE inhibited facultative melanogenesis in melanocytes
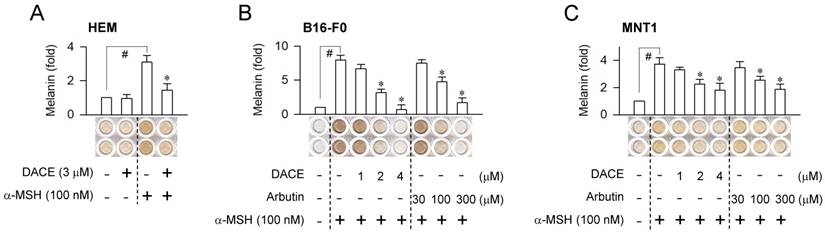
Melanogenic hormones such as α-MSH and ET-1 are secreted mainly by keratinocytes in UV-B-exposed skins [5]. They bind to the receptors MC1R and EDNRB on the membrane surface of melanocytes, which stimulates intracellular signaling pathways for melanin biosynthesis [5, 14]. We asked whether the in vivo outcomes of DACE could be translated into facultative melanogenesis in melanocytes, since DACE inhibits α-MSH-induced melanin pigmentation in B16-F0 cells [26]. Upon exposure to α-MSH alone, human epidermal melanocyte (HEM) or melanoma cell lines such as B16-F0 and MNT1 markedly increased intracellular and extracellular levels of melanin pigments (Figure 2A-C). Treatment with DACE significantly inhibited α-MSH-induced melanin production in HEM cells but did not affect the basal pigmentation (Figure 2A). DACE also inhibited melanin production in α-MSH-activated B16-F0 or MNT1 cells, as did arbutin, in which DACE showed more effectiveness (≥ 50-fold) than arbutin in the comparison with IC50 values (Figure 2B-C). Moreover, DACE inhibited ET-1-, N6,2'-O-dibutyryl (db)-cAMP- and WH-4-023-induced melanin production in B16-F0 cells (Figure S2A-C). db-cAMP was employed as a cell-permeable cAMP agonist and WH-4-023 as a SIK inhibitor, since α-MSH- and ET-1-activated melanocytes elevate intracellular cAMP levels followed by decrease in SIK activity, regulating the nuclear-cytoplasmic shuttling of CRTC1 via phosphorylation [1, 19]. However, DACE at the concentrations with antimelanogenic activity did not affect the viability or the proliferation of B16-F0 and MNT1 cells (Figure S3A-B), excluding nonspecific cytotoxicity. The results suggest that DACE could inhibit the melanogenic process downstream of SIK activity in cAMP-elevated melanocytes.
Effect of DACE on melanin production. (A) HEM cells were stimulated with α-MSH for 96 h in the presence of DACE. (B, C) B16-F0 or MNT1 cells were stimulated with α-MSH for 72 h in the presence of DACE. Melanin pigments were quantified by measuring absorbance values at 405 nm, and are represented as relative fold change. Data are mean ± SEM (n = 5). #P < 0.05 vs. medium alone. *P < 0.05 vs. α-MSH alone.
DACE suppressed the transcription of MITF-M and TYR
To understand the molecular basis of antimelanogenic action, we examined whether DACE could affect cellular levels of MITF-M, a master transcription factor that regulates melanogenesis and differentiation of cAMP-elevated melanocytes [9, 10]. Topical treatment with DACE decreased not only the protein levels of MITF-M (59 kDa) in UV-B-exposed skins, in which anti-MITF-M antibody recognized MITF-Ms in phosphorylated or unphosphorylated status (Figure 3A), but also its mRNA levels (Figure 3B). DACE also attenuated α-MSH-induced protein and mRNA levels of MITF-M in HEM cells (Figure 3C-D). Moreover, DACE decreased α-MSH-induced mRNA levels of MITF-M in MNT1 cells (Figure S4A), as well as α-MSH-, ET-1- or db-cAMP-induced mRNA levels of MITF-M in B16-F0 cells (Figure S4B). Transcriptional regulation of MITF-M by DACE was further determined using a promoter-dependent reporter assay. B16-F0 cells were transfected with MITF-M-Luc, a construct encoding the promoter region (-2200/+95) of MITF-M fused with firefly luciferase reporter. Upon exposure to α-MSH alone, B16-F0 cells harboring MITF-M-Luc displayed markedly higher luciferase activity (Figure 3E). Treatment with DACE inhibited the α-MSH-induced expression of luciferase, reporting promoter activity of MITF-M gene (Figure 3E). Thus, DACE could down-regulate the transcription of MITF-M.
Effect of DACE on MITF-M expression. (A, B) Dorsal skins of guinea pigs were exposed to UV-B radiation (350 mJ/cm2) and treated topically with DACE according to the protocol in Figure S1. Skin tissues were biopsied. HEM cells were pretreated with DACE for 2 h and stimulated with α-MSH for 4 h (C) or 2 h (D) in the presence of DACE. (A, C) Protein extracts were subjected to Western blot analysis (WB) with anti-MITF-M or anti-GAPDH antibody. (B, D) Total RNAs were subjected to RT-PCR analysis of MITF-M with the internal control β-actin. (E) B16-F0 cells were transfected with MITF-M-Luc reporter construct in combination with Renilla control vector. The transfected cells were pretreated with DACE for 2 h and stimulated with α-MSH for 20 h in the presence of DACE. Firefly luciferase activity, reporting promoter activity of MITF-M gene, is represented as relative fold change after normalizing to Renilla activity as a reference of transfection efficiency. B16-F0 cells were pretreated with DACE for 2 h, stimulated with α-MSH for 20 min (F, H) or 1 h (G) in the presence of DACE, and subjected to chromatin immunoprecipitation (ChIP) analysis. Chromatin fragments were precipitated with anti-CRTC1 (F), anti-SOX10 (G) or anti-β-catenin antibody (H). Input and precipitated DNAs were subjected to PCR encompassing CREB-responsive CRE motif (F), SOX10- (G) or LEF1-responsive cis-acting element (H) at the MITF-M promoter. Data are mean ± SEM (n = 3). #P < 0.05 vs. normal skin (A, B) or medium alone (C-H). *P < 0.05 vs. UV-B alone (Α, Β) or α-MSH alone (C-G).
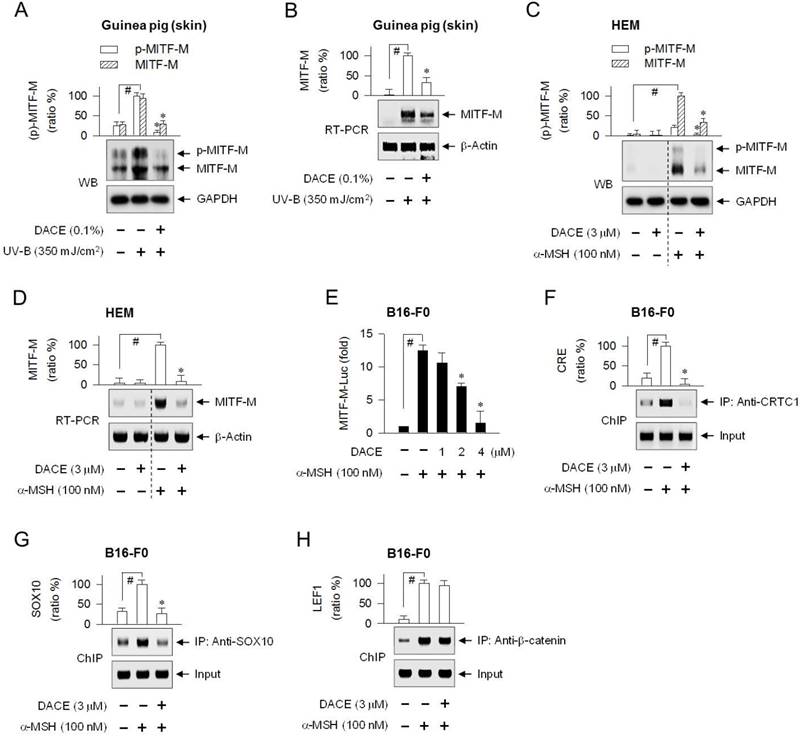
The MITF-M promoter contains a number of cis-acting elements, where the transcription machinery is positively or negatively regulated by transcription factors that are associated with signaling pathways in the melanogenic processes [28, 29]. Among the response elements that are potentially required to up-regulate MITF-M transcription in cAMP-elevated melanocytes, the CREB-responsive CRE motif is located at -291/-284 in the proximal region of mouse MITF-M promoter, the LEF1-binding sites at -366/-337, and the SOX10-responsive element at -426/406 (Figure S5A).
To determine which transcription factors or cis-acting elements are regulated by DACE, we performed chromatin immunoprecipitation (ChIP) assays. The CREB/CRTC1 heterodimer, SOX10, and LEF1/β-catenin heterodimer occupied the MITF-M promoter, and their specific binding was markedly increased upon exposure to α-MSH (Figure 3F-H; Figure S5B-D). Treatment with DACE decreased the occupancy of CREB/CRTC1 heterodimer at the CRE motif (-291/-284) of the MITF-M promoter in α-MSH-activated B16-F0 cells (Figure 3F; Figure S5B), as well as that of SOX10 at the cis-acting element (-426/-406), apart from the start site of MITF-M transcription (Figure 3G; Figure S5C). However, DACE exerted no effect on the α-MSH-enhanced binding of LEF1/β-catenin heterodimer to the promoter (Figure 3H; Figure S5D). The results suggest that DACE could suppress MITF-M expression by negatively regulating the transcriptional abilities of CREB/CRTC1 heterodimer and SOX10 but not that of LEF1/β-catenin heterodimer at the promoter.
MITF-M is a pivotal activator of the melanocyte-specific transcription of TYR and other melanogenic genes [9]. We examined whether DACE could affect cellular levels of TYR, a key enzyme in the biosynthetic pathway of melanin pigments [11]. Topical treatment with DACE decreased not only the protein levels of TYR (79 kDa) but also its mRNA levels in UV-B-exposed skins (Figure S6A-B). DACE also decreased the protein and mRNA levels of TYR in α-MSH-activated B16-F0 cells (Figure S6C-D). Moreover, DACE inhibited α-MSH-induced promoter activity of TYR, as determined using B16-F0 cells transfected with TYR-Luc, a construct encoding the promoter region (-2236/+59) of TYR fused with firefly luciferase reporter (Figure S6E). The results suggest that DACE could suppress the transcription of TYR, which could be attributed to its upstream effect on MITF-M.
DACE did not affect the phosphorylation circuits that act on CREB, CRTC1 and β-catenin
PKA directly phosphorylates CREB, SIK and β-catenin in cAMP-elevated melanocytes [1, 18, 30]. The phosphorylation of CREB plays an important role in the recruitment of CBP and p300 onto the chromatin-containing target promoter [1, 2]. p-SIK has lower kinase activity than dephospho-SIK in the phosphorylation of CRTC1 at Ser-151 and Ser-245 [18]. In turn, p-CRTC1 is sequestered in the cytoplasm by tethering with 14-3-3 proteins, while dephospho-CRTC1 shuttles across the nuclear envelope [19, 20]. p-β-catenin accumulates in the cytoplasm, due to its stable conformation from degradation, and allows it to be transported into the nucleus [30].
DACE had no effect on the ligation of the fluorescent α-MSH probe (TAMRA-NDP-α-MSH) with MC1R on the surface of B16-F0 cells (Figure S7A), excluding the possibility that DACE could antagonize the receptor or scavenge the ligand. We then examined whether DACE could affect the phosphorylation of CREB coupled with the dephosphorylation of CRTC1. The (de)phosphorylation circuits acting on CREB and CRTC1 are immediately upstream of MITF-M transcription in cAMP-elevated melanocytes [1]. Upon exposure to α-MSH alone, B16-F0 cells phosphorylated CREB (43 kDa) at Ser-133 (Figure S7B), and dephosphorylated CRTC1 (78 kDa) at Ser-151 (Figure S7C). Treatment with DACE affected neither the phosphorylation of CREB nor the dephosphorylation of CRTC1 in α-MSH-activated B16-F0 cells (Figure S7B-C). Furthermore, we performed an immunoprecipitation (IP) assay with a pan antibody against 14-3-3 proteins, and then probed these with anti-CRTC1 antibody to detect the co-precipitates. The CRTC1 was tethered with 14-3-3 (30 kDa) in the basal conditions of B16-F0 cells, but the protein assembly detached upon exposure to α-MSH (Figure S7D), synchronizing with the (de)phosphorylated status of CRTC1 (Figure S7C). Treatment with DACE showed no effect on the physical interaction between CRTC1 and 14-3-3 proteins (Figure S7D). α-MSH-activated B16-F0 cells contained β-catenin (92 kDa) that was phosphorylated at Ser-675, and concomitantly increased the protein levels, but not mRNA levels, of β-catenin (Figure 8A-B). Treatment with DACE affected neither the phosphorylation of β-catenin nor the protein levels of β-catenin in α-MSH-activated B16-F0 cells (Figure S8A). The results suggest that DACE did not affect MC1R-mediated signaling pathways in the phosphorylation of CREB and β-catenin, and those in the dephosphorylation of CRTC1.
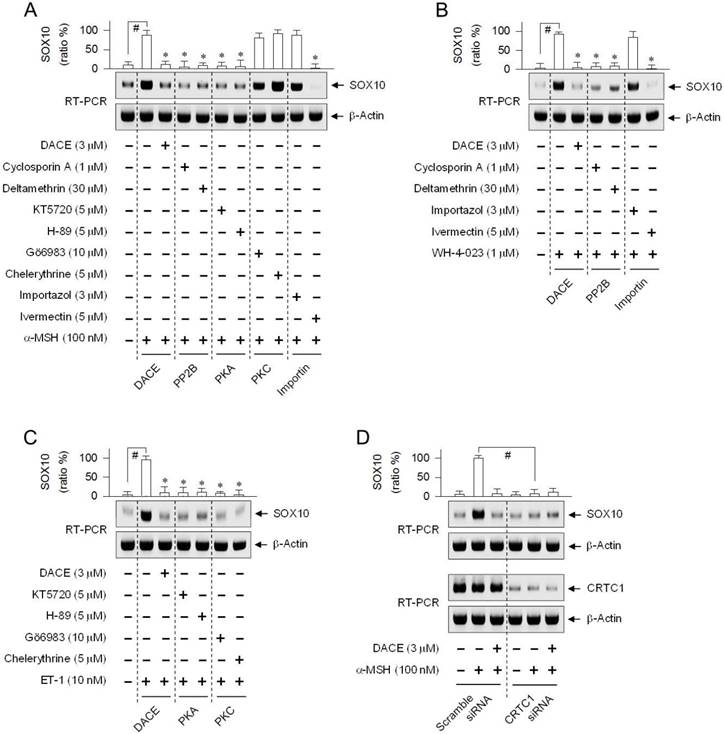
Effects of phosphatase inhibitor, kinase inhibitor and CRTC1-targeted siRNA on SOX10 induction. B16-F0 cells were pretreated with each inhibitor for 2 h and stimulated with α-MSH (A), WH-4-023 (B) or ET-1 (C) for 1 h in the presence of each inhibitor. (D) B16-F0 cells were transfected with scramble siRNA or CRTC1 siRNA. The transfected cells were stimulated with α-MSH for 1 h. Total RNAs were subjected to RT-PCR analysis of SOX10 with the internal control β-actin. Data are mean ± SEM (n = 3). #P < 0.05 vs. medium alone (A-C) or scramble siRNA (D). *P < 0.05 vs. α-MSH, WH-4-023 or ET-1 alone (A-C).
DACE blocked the nuclear import of CRTC1
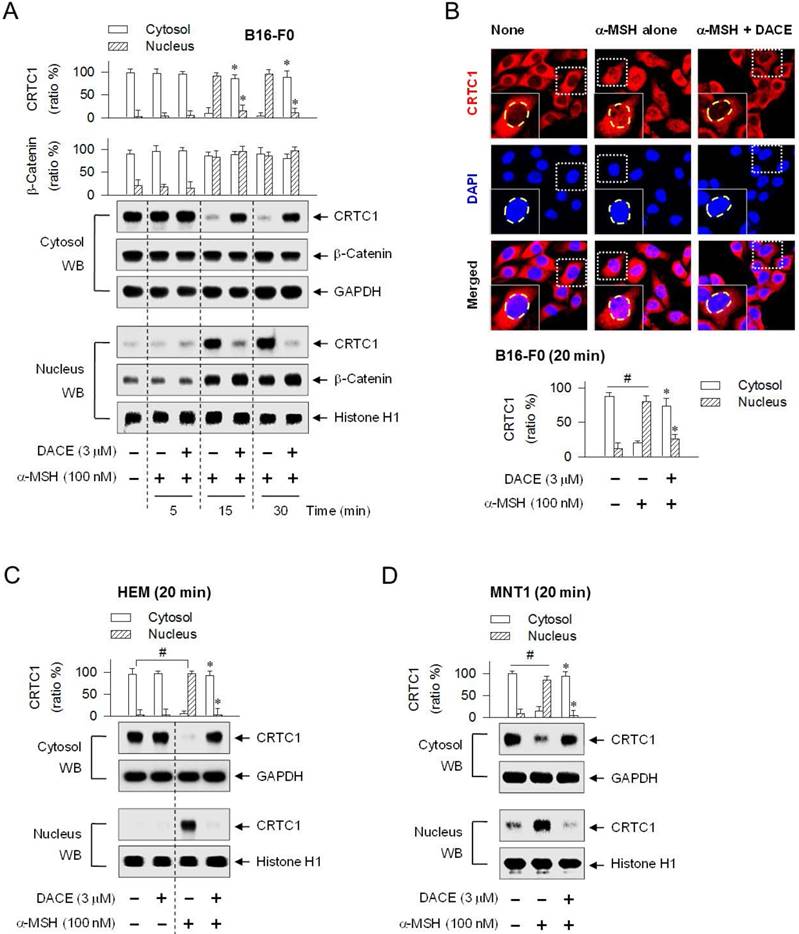
CREB was predominantly localized in the nucleus of B16-F0 cells under basal conditions or activated by α-MSH (Figure S9). Treatment with DACE had no effect on the nuclear retention of CREB (Figure S9). We then examined whether DACE could regulate the translocation of CRTC1 in and out of the nucleus, since DACE had decreased the occupancy of CREB/CRTC1 heterodimer at the MITF-M promoter in the nucleus (Figure 3F; Figure S5B) but had no effects on the dephosphorylation of CRTC1 and the physical tethering between CRTC1 and 14-3-3 proteins in the cytoplasm (Figure S7C-D). Upon exposure to α-MSH alone, B16-F0 cells shifted the localization of CRTC1 from the cytosol to the nucleus at the time points of 15 min to 1 h and exported CRTC1 out of the nucleus at 2 h, in which GAPDH (37 kDa) was used as a cytoplasmic marker and histone H1 (33 kDa) as a nucleus marker (Figure 4A; Figure S10A). Treatment with DACE inhibited the nuclear import of CRTC1 in α-MSH-activated B16-F0 cells (Figure 4A; Figure S10A). The mechanism of DACE, blocking entry of CRTC1 into the nucleus, was also confirmed in confocal fluorescence microscopy after staining CRTC1 as red and the nucleus as blue (Figure 4B). DACE consistently inhibited the nuclear entry of CRTC1 in α-MSH-activated HEM or MNT1 cells (Figure 4C-D). Moreover, DACE inhibited the nuclear entry of CRTC1 in ET-1- or WH-4-023-activated B16-F0 cells (Figure S10B-C). On the other hand, DACE exerted no effect on the nuclear retention of β-catenin in α-MSH-activated B16-F0 cells (Figure 4A).
Effect of DACE on the nuclear import of CRTC1. B16-F0 (A, B), HEM (C) or MNT1 cells (D) were pretreated with DACE for 2 h and stimulated with α-MSH for indicated time points in the presence of DACE. Cell extracts were partitioned between the cytosol and the nucleus, and subjected to Western blot analysis (WB) with anti-CRTC1, anti-β-catenin, anti-GAPDH or anti-histone H1 antibody. (B) The cells were subjected to confocal fluorescence microscopy. CRTC1 was stained red with anti-CRTC1 antibody followed by Alexa Fluor 594-labeled antibody and the nucleus as blue with DAPI. Data are mean ± SEM (n = 3). *P < 0.05 vs. α-MSH alone. #P < 0.05 vs. medium alone.
Nuclear-cytoplasmic shuttling of CRTC1 is regulated by its phosphorylation status [19]. Inhibition of PP2B or PKA restores p-CRTC1 levels in α-MSH- or db-cAMP-activated B16-F0 cells, whereas the inhibition of PP1, PP2A or p38 MAPK has no effects, even though all of them decrease melanin production [31]. We further examined which phosphatase or kinase pathway could switch the translocation of CRTC1 in α-MSH-, ET-1- and WH-4-023-activated cells. PP2B inhibitors (cyclosporin A, deltamethrin) and PKA inhibitors (KT5720, H-89) interrupted the nuclear import of CRTC1 in α-MSH-activated B16-F0 cells, but dual inhibitors of PP1 and PP2A (okadaic acid, calyculin A) and PKC inhibitors (Gö6983, chelerythrine) did not have significant effects on the nuclear import (Figure S11A-B). Moreover, the inhibitors of PP2B or PKA, in addition to the PKC inhibitors, blocked ET-1-induced entry of CRTC1 into the nucleus, while MEK1/2 inhibitors (PD98059, U0126) and an RSK inhibitor (SL0101) had no effects on the nuclear entry (Figure S11C). However, the PP2B inhibitors were only ones that blocked WH-4-023-induced nuclear import of CRTC1 (Figure S11D). The results suggest that PKA or PKC could function upstream of SIK activity, PKC might be involved in ENDRB- but not in MC1R-mediated signaling pathway, and PP2B could directly remove the phosphate groups on CRTC1 to induce nuclear translocation.
DACE down-regulated SOX10 induction
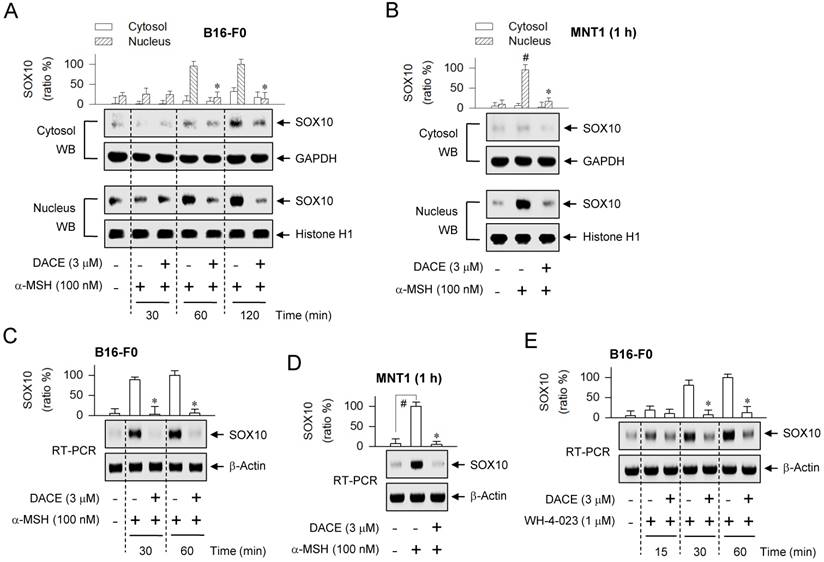
cAMP signaling through CREB/CRTC1 heterodimer, ubiquitous in almost all cell types, is adapted to the melanocyte-specific transcriptional activation of MITF-M in cooperation with SOX10 [22]. DACE had inhibited the occupancy of SOX10 at the MITF-M promoter (Figure 3G; Figure S5C). We examined whether DACE could also affect the nuclear retention of SOX10, as it had altered the nuclear import of CRTC1 (Figure 4A-D; Figure S10A-C). Upon exposure to α-MSH alone, B16-F0 cells markedly increased the nuclear levels of SOX10 (58 kDa) at the time points of 1-2 h (Figure 5A). Treatment with DACE suppressed the protein levels of SOX10 in the nucleus of α-MSH-activated B16-F0 or MNT1 cells, but its mechanism was not related to the translocation of SOX10 in and out of the nucleus (Figure 5A-B). Interestingly, either the stimulation of MC1R with α-MSH or the inhibition of SIK activity with WH-4-023 increased the mRNA levels of SOX10 (Figure 5C-E). Treatment with DACE attenuated α-MSH-induced mRNA levels of SOX10 in B16-F0 or MNT1 cells (Figure 5C-D), as well as those in WH-4-023-activated B16-F0 cells (Figure 5E). Thus, SOX10 would be inducible in the facultative melanogenesis. Moreover, DACE could down-regulate SOX10 induction, thereby resulting in an attenuated occupancy of SOX10 at the MITF-M promoter.
Effect of DACE on SOX10 induction. B16-F0 or MNT1 cells were pretreated with DACE for 2 h and stimulated with α-MSH (A-D) or WH-4-023 (E) for indicated time points in the presence of DACE. (A, B) Cell extracts were partitioned between the cytosol and the nucleus, and subjected to Western blot analysis (WB) with anti-SOX10, anti-GAPDH or anti-histone H1 antibody. (C-E) Total RNAs were subjected to RT-PCR analysis of SOX10 with the internal control β-actin. Data are mean ± SEM (n = 3). *P < 0.05 vs. α-MSH or WH-4-023 alone. #P < 0.05 vs. medium alone.
KPNA escorted CRTC1 into the nucleus
Small molecules or polypeptides less than about 40 kDa can passively move across the nuclear envelope, but larger proteins generally require the nuclear localizing sequences (NLSs) for entering into the nucleus [32, 33]. KPNA (also called importin-α) functions as an adaptor vehicle that recognizes the NLS on cargo protein, and then associates with KPNB (also called importin subunit β1) or other isoforms [32, 33]. Alternatively, many karyopherin-β members, such as transportins and importins, recognize their cargo protein without the help of KPNA [32, 33]. Cargo proteins loaded on KPNA or karyopherin-β family in the cytoplasm are actively transported through the nuclear envelope-localized nuclear pore complex (NPC) into the nucleus [32, 33].
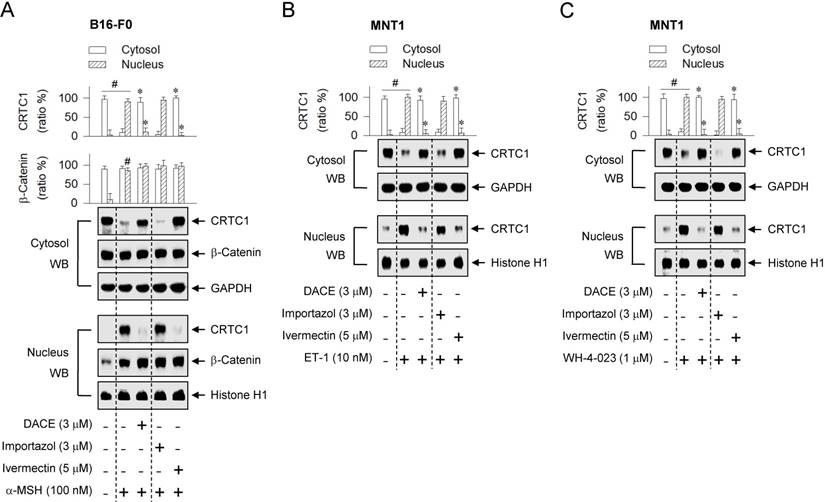
CRTC1 bears an Arg-rich NLS [4, 34], but the molecular basis of its nuclear translocation remains unknown. To determine whether CRTC1 would be escorted into the nucleus by the classical NLS- dependent machinery, ivermectin was employed as a KPNA inhibitor, and importazol as a KPNB inhibitor [35, 36]. Treatment with ivermectin, but not with importazol, blocked the nuclear entry of CRTC1 in α-MSH-activated B16-F0 cells (Figure 6A), even though both inhibited melanin production (Figure S12). Ivermectin consistently interrupted the nuclear import of CRTC1 in ET-1- or WH-4-023-activated MNT1 cells, while importazol had no effect on the nuclear import (Figure 6B-C). On the other hand, neither ivermectin nor importazol inhibited the nuclear retention of β-catenin in α-MSH-activated B16-F0 cells (Figure 6A). The results suggest that CRTC1 would be escorted into the nucleus by ivermectin-sensitive KPNA subtypes.
Effects of importazol and ivermectin on the nuclear import of CRTC1. B16-F0 or MNT1 cells were pretreated with importazol or ivermectin for 2 h and stimulated with α-MSH (A), ET-1 (B) or WH-4-023 (C) for 20 min in the presence of importazol or ivermectin. Cell extracts were partitioned between the cytosol and the nucleus, and subjected to Western blot analysis (WB) with anti-CRTC1, anti-β-catenin, anti-GAPDH or anti-histone H1 antibody. Data are mean ± SEM (n = 3). #P < 0.05 vs. medium alone. *P < 0.05 vs. α-MSH, ET-1 or WH-4-023 alone.
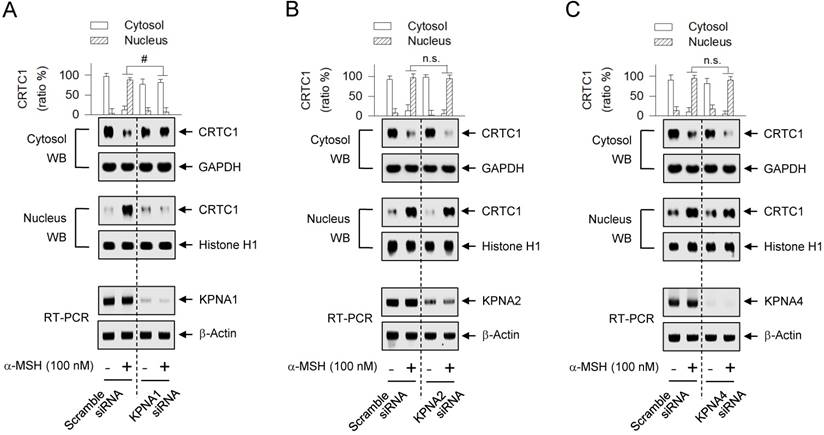
The human genome encodes seven isoforms of KPNA that are classified into three subfamilies; i) KPNA1 (also called Impα5), KPNA5 (Impα6) and KPNA6 (Impα7), ii) KPNA2 (Impα1) and KPNA7 (Impα8), and iii) KPNA3 (Impα4) and KPNA4 (Impα3) [33, 37]. KPNA subtypes recognize their cargo protein with remarkable specificity [37]. To examine which KPNA subtype could escort CRTC1 as a cargo across the nuclear envelope, we employed a gene-specific siRNA-based approach. Knockdown of KPNA1 led to a significantly decreased translocation of CRTC1 from the cytosol to the nucleus in α-MSH-activated MNT1 cells (Figure 7A), while siRNAs targeting KPNA2 and KPNA4 did not show significant effects (Figure 7B-C). This is the first report that specific KPNA subtype (KPNA1) could recognize the Arg-rich NLS on CRTC1 and escort it into the nucleus.
Effect of KPNA subtype-targeted siRNA on the nuclear import of CRTC1. MNT1 cells were transfected with KPNA1 siRNA (A), KPNA2 siRNA (B), KPNA4 siRNA (C) or scramble siRNA (A-C). The transfected cells were stimulated with α-MSH for 20 min. Cell extracts were partitioned between the cytosol and the nucleus, and subjected to Western blot analysis (WB) with anti-CRTC1, anti-GAPDH or anti-histone H1 antibody. Data are mean ± SEM (n = 3). #P < 0.05 vs. scramble siRNA. Abbreviation; n.s., not significant.
SOX10 induction was regulated by CRTC1
DACE had blocked the nuclear import of CRTC1 (Figure 4A-D; Figure S10A-C) and inhibited the induction of SOX10 (Figure 5A-E), in which melanocytic-lineage cells transported CRTC1 from the cytosol to the nucleus at 15-20 min after stimulation with α-MSH, ET-1 or WH-4-023, and increased the mRNA and protein levels of SOX10 at 30 min to 1 h. We examined whether the inhibitors of PP2B, PKA, PKC, KPNA or KPNB could also affect SOX10 induction, since they had altered the nuclear import of CRTC1 (Figure 6A-C; Figure S11A-D). The PP2B inhibitors (cyclosporin A, deltamethrin) and the PKA inhibitors (KT5720, H-89) attenuated α-MSH-induced SOX10 induction in B16-F0 cells, while the PKC inhibitors (Gö6983, chelerythrine) did not have significant effects (Figure 8A). Interestingly, the KPNA inhibitor (ivermectin), but not the KPNB inhibitor (importazol), decreased SOX10 induction in α-MSH- and WH-4-023-activated cells (Figure 8A-B). The inhibition of PP2B also attenuated SOX10 induction in WH-4-023-activated cells (Figure 8B), and the inhibition of PKA or PKC decreased SOX10 induction in ET-1-activated cells (Figure 8C). Thus, effects of pharmacological agents on SOX10 induction were coincident whether or not they primarily inhibited the nuclear import of CRTC1. This indicates that SOX10 induction might be coupled with the nuclear-cytoplasmic shuttling of CRTC1.
To test the proposed cross-talk between CRTC1 and SOX10, we employed a gene-specific siRNA-based approach. In B16-F0 cells harboring CRTC1-targeted siRNA, the mRNA levels of CRTC1 were severely depleted, irrespective of absence or presence of α-MSH and DACE, as compared with those in the control cells transfected with scramble siRNA (Figure 8D). Upon exposure to α-MSH, control cells increased the mRNA levels of SOX10 as expected, while knockdown of CRTC1 led to a significant decrease in the amount of SOX10 transcript (Figure 8D). The results indicate, for the first time, that SOX10 induction would be at least partially controlled by CRTC1.
Discussion
The aberrant production and distribution of melanin pigments cause skin disorders with heavily pigmented patches, such as melasma, freckles and senile lentigo [5, 8]. The hit compounds, regulating the synthesis of melanin biopolymers or the transfer of pigmented melanosomes, have been identified in cell-based assays [38, 39]. However, a large numbers of the antimelanogenic candidates have met with limited or no success in the cutaneous treatment of patients, suggesting that clinical effectiveness must overcome the challenge penetrating the skin barrier [39]. The skin of guinea pig displays similar kinetic parameters to human skin in the transdermal absorption of various chemicals, thus can be used as a surrogate for human skin in studies requiring cutaneous application [40].
In the current study, topical treatment with DACE prevented UV-B-induced hyperpigmentation in the skin with a concomitant decrease in the mRNA and protein levels of MITF-M and TYR. DACE also inhibited melanin production in α-MSH- or ET-1-activated HEM, B16-F0 and MNT1 cells. The melanocytic-lineage cells transported CRTC1 from the cytosol to the nucleus at 15-20 min after the stimulation of MC1R with α-MSH or that of ENDRB with ET-1, and increased the mRNA and protein levels of SOX10 at 30 min to 1 h, MITF-M at 1-4 h or TYR at 18-48 h, thus leading to heavy pigmentation with melanin biopolymers at 72-96 h.
DACE directly blocked the nuclear import of CRTC1 in melanocytes. DACE resultantly inhibited SOX10 induction, and suppressed the transcriptional abilities of CREB/CRTC1 heterodimer and SOX10 at the MITF-M promoter. However, DACE did not affect the nuclear retention of CREB, the phosphorylation of CREB, the dephosphorylation of CRTC1, and the physical tethering between CRTC1 and 14-3-3 proteins. Based on these insights, we propose a mechanism of DACE on the antimelanogenic action (Figure 9).
A proposed mechanism of DACE on antimelanogenic action. DACE blocked the nuclear import of CRTC1 in melanocytes. This led to the inhibition of SOX10 induction, and the suppression in the transcriptional abilities of CREB/CRTC1 heterodimer and SOX10 at the MITF-M promoter, MITF-M is a master regulator in the expression of TYR or other melanogenic genes. Abbreviation; AC, adenylate cyclase.
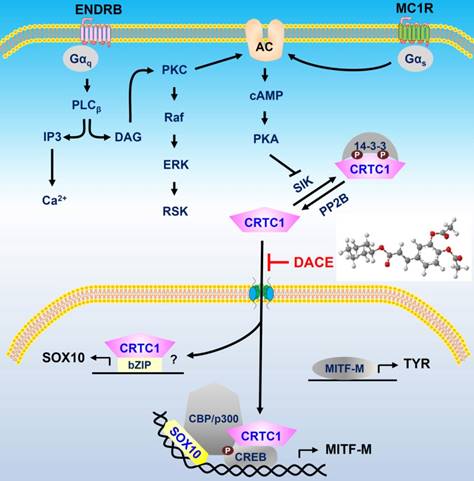
B16 cells express CRTC1 at a higher level than CRTC2 and CRTC3 [41]. Overexpression of the dominant-negative CRTC1 inhibits UV-B-induced melanogenesis in B16 cells, while overexpression of CRTC1 up-regulates the mRNA levels of MITF-M with a concomitant increase in the production of melanin pigments [41]. In the current study, we discovered CRTC1 as a druggable target in acquired pigmentary disorder of the skin. DACE blocked the nuclear entry of CRTC1 in α-MSH-, ET-1- or WH-4-023-activated melanocytes, as did ivermectin. However, DACE had no effects on the nuclear import of CREB or β-catenin in α-MSH-activated B16-F0 cells. Ivermectin also did not affect the nuclear retention of β-catenin. Ivermectin, an FDA-approved anti-parasitic drug, masks the NLS-binding pocket of KPNA that is needed for the recognition of cargo proteins across the nuclear envelope [35]. CREB requires KPNB alone, but not KPNA, in the nuclear import [42]. β-Catenin moves from the cytosol to the nucleus by binding directly to NPC components, independent of the NLS or karyopherin-β members [43].
CRTCs have similar modular structures, consisting of an Arg-rich NLS and two Leu-rich nuclear export sequences (NESs) [4, 19]. XPO1, also called exportin 1 or chromosome maintenance 1 (CRM1), exports CRTCs from the nucleus [19]. XPO1 recognizes the Leu-rich NESs, and its inhibition sequesters CRTC1 in the nucleus [19]. However, little is known about nuclear import of CRTC1. The Arg-rich NLS on CRTC1 is essential for its nuclear entry. Two mutants on the NLS region, Ala substitutions in place of the four Arg residues at positions 103, 106, 108 and 110 or another Ala substitutions instead of the Pro at position 114 and the two Arg residues at positions 116 and 117, impair its nuclear translocation [34]. In the current study, knockdown of KPNA1 led to a significantly decrease in the nuclear import of CRTC1 in α-MSH-activated MNT1 cells. Taken together, DACE could inhibit either the function of NLS of CRTC1 or the escorting of CRTC1 across the nuclear envelope by ivermectin-sensitive KPNA subtype such as KPNA1.
Small-molecule SIK inhibitors increase melanin pigmentation in the dorsal skins of MC1R-deficient mice, and promote epidermal darkening in human breast skin explants [44]. Overexpression of SIK counteracts the expression of MITF-M and the production of pigmented melanosomes in CRTC1-overexpressed B16 cells [41]. In the current study, the inhibition of SIK activity with WH-4-023 increased the mRNA and protein levels of SOX10, following the nuclear import of CRTC1, in melanocytic-lineage cells. This was also observed when MC1R was stimulated with α-MSH, or when ENDRB was stimulated with ET-1. Knockdown of CRTC1 led to a significant decrease in SOX10 induction in α-MSH-activated B16-F0 cells. DACE blocked the nuclear import of CRTC1, and then coupled to the inhibition of SOX10 induction in α-MSH-, ET-1- or WH-4-023-activated cells. However, the molecular basis of CRTC1-directed SOX10 induction remains to be clarified. Recently, the role of CRTCs has been expanded beyond CREB [4]. CRTCs co-activate other bZIP transcription factors, including AP-1 and ATF6, but compete with ATF3 and farnesoid X receptor in the occupancy at target promoters [4]. CRTCs also serve as scaffolds in the assembly of spliceosome components, excising introns from pre-mRNA transcripts [45].
In conclusion, DACE mitigated UV-B-induced hyperpigmentation in the skin by blocking the nuclear import of CRTC1 in melanocytes. This unique mechanism led to the inhibition of SOX10 induction, and the suppression in the transcriptional abilities of CREB/CRTC1 heterodimer and SOX10 at the MITF-M promoter, thus preventing facultative melanogenesis. Furthermore, current study is the first to report that i) KPNA1 could escort CRTC1 as a cargo across the nuclear envelope, ii) SOX10 would be inducible in the melanogenic process, and iii) CRTC1 could direct SOX10 induction at the transcription level. Therefore, we propose the nuclear import of CRTC1 as a druggable target in the treatment of acquired pigmentary disorders such as melasma, freckles and senile lentigo.
Materials and Methods
Drugs and chemicals
DACE (> 99% purity) was synthesized as previously described [26]. The pharmacological agents used in this study were arbutin (Sigma- Aldrich, A4256) as a skin whitener; WH-4-023 (Sigma- Aldrich, SML1334) as a SIK inhibitor; importazol (Sigma-Aldrich, SML0341) as a KPNB inhibitor; ivermectin (Sigma-Aldrich, I8898) as a KPNA inhibitor; okadaic acid (Sigma-Aldrich, O9381) and calyculin A (Sigma-Aldrich, C5552) as dual inhibitors of PP1 and PP2A; cyclosporin A (Sigma-Aldrich, C1822) and deltamethrin (Sigma-Aldrich, D9315) as PP2B inhibitors; KT5720 (Sigma-Aldrich, K3761) and H-89 (Sigma-Aldrich, B1427) as PKA inhibitors; Gö6983 (Sigma-Aldrich, G1918) and chelerythrine (Sigma-Aldrich, C2932) as PKC inhibitors; PD98059 (Sigma-Aldrich, P215) and U0126 (Sigma-Aldrich, U120) as MEK1/2 inhibitors; and SL0101 (Merck, 559285) as an RSK inhibitor.
UV-B-induced skin hyperpigmentation
Brownish guinea pigs (male, 6-7 weeks old) were purchased from Daehan Biolink (Eumsung, Korea), and housed in the facility at temperature (22 ± 2℃), with humidity (55 ± 5%) and a 12 h/12 h light-dark cycle under free access to food and water ad libitum. After they were shaved, the dorsal skin of each guinea pig was divided into four areas (2 cm x 2 cm each). DACE was dissolved in a vehicle (propylene glycol: ethanol: H2O = 5: 3: 2). As shown in Figure S1, allocated skin areas were topically treated with DACE (50 μl each) in a twice-daily regimen for four consecutive weeks and irradiated with UV-B (350 mJ/cm2) once every two days for three consecutive weeks. Skin hyperpigmentation was measured at one week after the end of the UV-B radiation by scanning the pigmented area treated topically with vehicle or DACE. Skin tissues were fixed in 10% formaldehyde (Sigma-Aldrich, F8775), embedded in paraffin (Sigma-Aldrich, 327212), sectioned with a thickness of 5 μm, and stained with Fontana-Masson silver nitrate (Scytek, FMS-1) to examine melanin granules. Protein or RNA extracts were prepared from fresh tissues and subjected to Western blot analysis or reverse transcription-PCR (RT-PCR) analysis. The protocols were approved by the Animal Experimentation Ethics Committee in Chungbuk National University (permit number CBNUA-809-15-01), and conducted in accordance with the Korean Ministry of Food and Drug Safety Guide for the Care and Use of Laboratory Animals.
Cell culture
HEM cells (Thermo Fischer Scientific, C1025C) were cultured in medium 254 (GIBCO, M254) containing human melanocyte growth supplement (GIBCO, S002). B16-F0 mouse melanoma cell line (ATCC, CRL-6322) was cultured in DMEM (Sigma-Aldrich, D2902) containing 10% fetal bovine serum (FBS, Corning, 35-015-CV) and antibiotic-antimycotic cocktail (GIBCO, 15240062). MNT1 human melanoma cell line (a gift from Dr. E.-S. Oh at Ewha Womans University) was cultured in MEM (Welgene, LM007-7) containing 10% DMEM, 20% FBS, 20 mM HEPES (Sigma-Aldrich, H4034) and antibiotic-antimycotic cocktail. The cells were incubated under an atmosphere of 5% CO2 at 37℃.
Melanin quantification
HEM cells were stimulated with 100 nM α-MSH (Sigma-Aldrich, M4125) for 96 h. B16-F0 or MNT1 cells were stimulated with 100 nM α-MSH, 10 nM ET-1 (Sigma-Aldrich, E7764), 3 mM db-cAMP (Sigma-Aldrich, D0627) or 1 μM WH-4-023 for 72 h. Extracellular melanin in culture supernatants and intracellular melanin in cell pellets were disrupted in 0.85 N NaOH and 20% dimethyl sulfoxide (DMSO, Sigma-Aldrich, 34869) under heating at 80℃, and measured absorbance values at 405 nm.
Cell viability assay
B16-F0 cells were incubated with DACE for 72 h in the presence of α-MSH. The cells were reacted with 0.5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich, M5655) for 1 h. Formazan crystals were dissolved in 99% DMSO, and measured absorbance values at 590 nm. MNT1 cells were incubated with DACE for 72 h in the presence of α-MSH. Cell numbers were counted after exclusion with trypan blue dye (Sigma-Aldrich, T6146).
Western blot analysis
Protein extracts were prepared with RIPA buffer (iNtRON, IBS-BR001), resolved on SDS-acrylamide gels by electrophoresis and transferred to polyvinylidene difluoride membrane (Roche, 03010040001). Western blots were blocked with either 5% non-fat milk (Becton-Dickinson, 23100) or 5% bovine serum albumin (BSA, Affymetrix, 10857) in Tris-buffered saline containing 0.05% Tween 20 (Sigma-Aldrich, P1379). After washing, blots were reacted with primary antibody at 4℃ overnight followed by secondary antibody for 1 h at room temperature. Immune complex on the blots was visualized by reacting with enhanced chemiluminescence reagent (GE Healthcare, RPN2232). This study employed primary antibodies against MITF-M (Abcam, ab12039), TYR (Santa Cruz, sc-7833), p-CREB (Cell Signaling, 9198), CREB (Cell Signaling, 9197), p-CRTC1 (Cell Signaling, 3359), CRTC1 (Cell Signaling, 2587), 14-3-3 (Santa Cruz, sc-1657), p-β-catenin (Cell Signaling, 9567), β-catenin (Cell Signaling, 9562), SOX10 (Santa Cruz, sc-17342), GAPDH (Santa Cruz, sc-25778) or histone H1 (Santa Cruz, sc-8030). Secondary antibodies were rabbit anti-goat IgG labeled with horseradish peroxidase (HRP) (Thermo Fisher Scientific, A27011), goat anti-rabbit IgG labeled with HRP (Thermo Fisher Scientific, 31460), and goat anti-mouse IgG labeled with HRP (Thermo Fisher Scientific, 31430).
RT-PCR analysis
Total RNAs were prepared with NucleoZOL kit (Macherey-Nagel, 1609), and subjected to RT-PCR analysis in the determination of mRNA levels of MITF-M, TYR, β-catenin, SOX10, KPNA subtype or CRTC1 with the internal control β-actin. Nucleotide sequences of RT-PCR primers are described in Table S1. Briefly, total RNAs were reversely transcribed for 1 h at 42℃ with oligo-dT as a primer (iNtRON, 25087). Single-stranded cDNAs were subjected to 27-40 cycles of PCR using a premix kit (Bioneer, K2018), in which one cycle consisted of denaturation for 15-30 s at 95℃, annealing for 15-30 s at 54-60℃, and DNA extension for 25-60 s at 72℃. RT-PCR products were resolved on agarose gels by electrophoresis and stained with EcoDye (Biofact, ES301-1000).
Firefly luciferase reporter assay
B16-F0 cells were transfected with each reporter construct, MITF-M (-2200/+95)-Luc or TYR (-2236/+59)-Luc, in combination with Renilla control vector for 24 h using a lipofectamine kit (Invitrogen, 11668). The transfected cells were stimulated with 100 nM α-MSH for 20 h. Cell extracts were subjected to dual luciferase assay (Promega, E1910). Firefly luciferase activity, reporting promoter activity of MITF-M or TYR gene, was normalized to Renilla luciferase activity as a reference of transfection efficiency.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed according to the protocol supplied with a premix kit (Merck, 17-295). B16-F0 cells were reacted with 1% formaldehyde for 10 min to cross-link between DNA and proteins. Cell extracts were sonicated to yield chromatin fragments with about 200-500 base pairs (bp) in size. Chromatin fragments were incubated with anti-CRTC1 (Cell Signaling, 2587), anti-β-catenin (Cell Signaling, 9562) or anti-SOX10 antibody (Santa Cruz, sc-17342) at 4℃ overnight, and precipitated with protein A-sepharose bead-sheared salmon sperm slurry for 4 h. To reverse the cross-links, ChIP elutes were incubated with 80 mM NaCl for 6 h at 65℃, and supplemented with 40 mM Tris-HCl containing 0.4 mg/ml proteinase K (Sigma-Aldrich, P2308) and 10 mM EDTA in another incubation for 1 h at 45℃. Input and precipitated DNAs were subjected to quantitative PCR encompassing CREB-, LEF1- or SOX10-responsive element at the MITF-M promoter in mice. Nucleotide sequences of PCR primers are described in Table S2. Quantitative PCR with SYBR green (Qiagen 204054) was subjected to 40 cycles in a condition of denaturation for 10 s at 95℃ and combined annealing-DNA extension for 30 s at 60℃.
Flow cytometric analysis
B16-F0 cells were incubated with 500 nM TAMRA-NDP-α-MSH (Anasper, AS-60514-1), a fluorescent α-MSH probe, for 30 min at room temperature. After washing, cells were subjected to flow cytometric analysis in the determination of TAMRA-NDP-α-MSH ligation to the receptor.
Immunoprecipitation (IP)
Cell extracts were incubated with a pan antibody against 14-3-3 proteins (Santa Cruz, sc-1657) at 4℃ overnight, and precipitated with protein G-sepharose bead (GE Healthcare, 17-0618-01) for 4 h.
Confocal microscopy
B16-F0 cells were seeded onto poly Lys-coated slides, fixed in 4% formaldehyde, and blocked with phosphate-buffered saline containing 1% BSA, 0.1% gelatin (Sigma-Aldrich, G7041), 0.3% Triton X-100 (Sigma-Aldrich, X100), 0.05% Tween 20 and 0.05% sodium azide (Sigma-Aldrich, S2002). After washing, cells on the slides were reacted with anti-CRTC1 antibody (Cell Signaling, 2587) at 4℃ overnight followed by Alexa Fluor 594-labeled secondary antibody (Invitrogen, A21207) for 1 h, and also stained with 4',6-diamidino-2-phenylindole (DAPI, Vector, H-1200). CRTC1 was examined as a red color and nuclei as a blue color under confocal fluorescence microscope.
siRNA transfection
For endogenous silencing of specific genes, B16-F0 cells were transfected with KPNA subtype- or CRTC1-targeted siRNA for 48 h using a lipofectamine kit. Nucleotide sequences of siRNAs are described in Table S3.
Statistical analysis
Results are expressed as mean ± SEM. Data were analyzed with ANOVA followed by the Student's t-test. P values less than 0.05 were considered as statistically significant.
Acknowledgements
This work was financially supported by grants (2016R1A6A3A11933508, 2018R1D1A1B07045596) or MRC program (2017R1A5A2015541) from the National Research Foundation of Korea, by a grant (WISET 2017-519) from the Korean Ministry of Science and ICT, and by the foresting project of Osong academy-industry convergence from the Korean Ministry of Trade, Industry & Energy.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141-51
2. Solt I, Magyar C, Simon I. et al. Phosphorylation-induced transient intrinsic structure in the kinase-inducible domain of CREB facilitates its recognition by the KIX domain of CBP. Proteins. 2006;64:749-57
3. Conkright MD, Canettieri G, Screaton R. et al. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413-23
4. Escoubas CC, Silva-Garcia CG, Mair WB. Deregulation of CRTCs in aging and age-related disease risk. Trends Genet. 2017;33:303-21
5. Bastonini E, Kovacs D, Picardo M. Skin pigmentation and pigmentary disorders: focus on epidermal/dermal cross-talk. Ann Dermatol. 2016;28:279-89
6. Tadokoro R, Takahashi Y. Intercellular transfer of organelles during body pigmentation. Curr Opin Genet Dev. 2017;45:132-8
7. Mohania D, Chandel S, Kumar P. et al. Ultraviolet radiations: skin defense-damage mechanism. Adv Exp Med Biol. 2017;996:71-87
8. Yamaguchi Y, Hearing VJ. Melanocytes and their diseases. Cold Spring Harb Perspect Med. 2014;4:a017046
9. Vachtenheim J, Borovansky J. "Transcription physiology" of pigment formation in melanocytes: central role of MITF. Exp Dermatol. 2010;19:617-27
10. Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest. 2017;97:649-56
11. Rzepka Z, Buszman E, Beberok A. et al. From tyrosine to melanin: signaling pathways and factors regulating melanogenesis. Postepy Hig Med Dosw (Online). 2016;70:695-708
12. Haq R, Fisher DE. Targeting melanoma by small molecules: challenges ahead. Pigment Cell Melanoma Res. 2013;26:464-9
13. Shtivelman E, Davies MQ, Hwu P. et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5:1701-52
14. Swope VB, Abdel-Malek ZA. Significance of the melanocortin 1 and endothelin B receptors in melanocyte homeostasis and prevention of sun-induced genotoxicity. Front Genet. 2016;7:146
15. Rodriguez CI, Setaluri V. Cyclic AMP (cAMP) signaling in melanocytes and melanoma. Arch Biochem Biophys. 2014;563:22-7
16. Mountjoy KG, Kong PL, Taylor JA. et al. Melanocortin receptor-mediated mobilization of intracellular free calcium in HEK293 cells. Physiol Genomics. 2001;5:11-9
17. Hagiwara M, Brindle P, Harootunian A. et al. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol. 1993;13:4852-9
18. Uebi T, Tamura M, Horike N. et al. Phosphorylation of the CREB-specific coactivator TORC2 at Ser(307) regulates its intracellular localization in COS-7 cells and in the mouse liver. Am J Physiol Endocrinol Metab. 2010;299:E413-25
19. Bittinger MA, McWhinnie E, Meltzer J. et al. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156-61
20. Sonntag T, Vaughan JM, Montminy M. 14-3-3 proteins mediate inhibitory effects of cAMP on salt-inducible kinases (SIKs). FEBS J. 2018;285:467-80
21. Harris ML, Baxter LL, Loftus SK. et al. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010;23:496-513
22. Huber WE, Price ER, Widlund HR. et al. A tissue-restricted cAMP transcriptional response: SOX10 modulates α-melanocyte-stimulating hormone-triggered expression of microphthalmia-associated transcription factor in melanocytes. J Biol Chem. 2003;278:45224-30
23. Urtatiz O, Van Raamsdonk CD. Gnaq and Gna11 in the endothelin signaling pathway and melanoma. Front Genet. 2016;7:59
24. Sato-Jin K, Nishimura EK, Akasaka E. et al. Epistatic connections between microphthalmia-associated transcription factor and endothelin signaling in Waardenburg syndrome and other pigmentary disorders. FASEB J. 2008;22:1155-68
25. Cronin JC, Loftus SK, Baxter LL. et al. Identification and functional analysis of SOX10 phosphorylation sites in melanoma. PLoS One. 2018;13:e0190834
26. Jo H, Choi M, Sim J. et al. Synthesis and biological evaluation of caffeic acid derivatives as potent inhibitors of α-MSH-stimulated melanogenesis. Bioorg Med Chem Lett. 2017;27:3374-7
27. Maeda K, Fukuda M. Arbutin: mechanism of its depigmenting action in human melanocyte culture. J Pharmacol Exp Ther. 1996;276:765-9
28. Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015;72:1249-60
29. Shibahara S, Takeda K, Yasumoto K. et al. Microphthalmia-associated transcription factor (MITF): multiplicity in structure, function, and regulation. J Investig Dermatol Symp Proc. 2001;6:99-104
30. Bellei B, Pitisci A, Catricala C. et al. Wnt/β-catenin signaling is stimulated by α-melanocyte-stimulating hormone in melanoma and melanocyte cells: implication in cell differentiation. Pigment Cell Melanoma Res. 2011;24:309-25
31. Yun CY, Mi Ko S, Pyo Choi Y. et al. α-Viniferin improves facial hyperpigmentation via accelerating feedback termination of cAMP/PKA-signaled phosphorylation circuit in facultative melanogenesis. Theranostics. 2018;8:2031-43
32. Mahipal A, Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther. 2016;164:135-43
33. Cagatay T, Chook YM. Karyopherins in cancer. Curr Opin Cell Biol. 2018;52:30-42
34. Ch'ng TH, DeSalvo M, Lin P. et al. Cell biological mechanisms of activity-dependent synapse to nucleus translocation of CRTC1 in neurons. Front Mol Neurosci. 2015;8:a00048
35. Wagstaff KM, Sivakumaran H, Heaton SM. et al. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J. 2012;443:851-6
36. Soderholm JF, Bird SL, Kalab P. et al. Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem Biol. 2011;6:700-8
37. Pumroy RA, Cingolani G. Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem J. 2015;466:13-28
38. Gillbro JM, Olsson MJ. The melanogenesis and mechanisms of skin-lightening agents: existing and new approaches. Int J Cosmet Sci. 2011;33:210-21
39. Solano F, Briganti S, Picardo M. et al. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res. 2006;19:550-71
40. Barbero AM, Frasch HF. Pig and guinea pig skin as surrogates for human in vitro penetration studies: a quantitative review. Toxicol In Vitro. 2009;23:1-13
41. Horike N, Kumagai A, Shimono Y. et al. Downregulation of SIK2 expression promotes the melanogenic program in mice. Pigment Cell Melanoma Res. 2010;23:809-19
42. Forwood JK, Lam MH, Jans DA. Nuclear import of Creb and AP-1 transcription factors requires importin-b 1 and Ran but is independent of importin-α. Biochemistry. 2001;40:5208-17
43. Fagotto F, Gluck U, Gumbiner BM. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of β-catenin. Curr Biol. 1998;8:181-90
44. Mujahid N, Liang Y, Murakami R. et al. A UV-independent topical small-molecule approach for melanin production in human skin. Cell Rep. 2017;19:2177-84
45. Amelio AL, Miraglia LJ, Conkright JJ. et al. A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc Natl Acad Sci U S A. 2007;104:20314-9
Author contact
![]() Corresponding author: Youngsoo Kim, Ph.D. & Professor. College of Pharmacy, Chungbuk National University, Cheongju 28160, Korea. E-mail: youngsooac.kr. Tel: +82-43-261-2823. Fax: +82-43-268-2732
Corresponding author: Youngsoo Kim, Ph.D. & Professor. College of Pharmacy, Chungbuk National University, Cheongju 28160, Korea. E-mail: youngsooac.kr. Tel: +82-43-261-2823. Fax: +82-43-268-2732