Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2019; 9(15):4375-4390. doi:10.7150/thno.33688 This issue Cite
Research Paper
Autophagy impairment contributes to PBDE-47-induced developmental neurotoxicity and its relationship with apoptosis
Pei Li1,2,*, Rulin Ma1,2,*, Lixin Dong1,2, Luming Liu1,2, Guoyu Zhou1,2, Zhiyuan Tian1,2, Qian Zhao1,2, Tao Xia1,2, Shun Zhang1,2, ![]() , Aiguo Wang1,2,
, Aiguo Wang1,2, ![]()
1. Department of Occupational and Environmental Health, School of Public Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, People's Republic of China
2. Key Laboratory of Environment and Health, Ministry of Education & Ministry of Environmental Protection, State Key Laboratory of Environmental health (incubating), School of Public Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, People's Republic of China
* These authors contributed equally to this work.
Received 2019-1-31; Accepted 2019-5-29; Published 2019-6-9
Abstract

Apoptosis is involved in 2,2',4,4'- tetrabromodiphenyl ether (PBDE-47)-induced developmental neurotoxicity. However, little is known about the role of autophagy, especially its relationship with apoptosis underlying such neurotoxic process.
Methods: Using female Sprague-Dawley rats exposed to low-dose PBDE-47 (0.1, 1.0 and 10 mg/kg/day) from pre-pregnancy until weaning of offspring to mimic human exposure, we investigated the effects of PBDE-47 on autophagy and apoptosis in relation to cognitive impairment of adult offspring rats. We also evaluated relationship between autophagy and apoptosis using neuroendocrine pheochromocytoma (PC12) cells, a widely used neuron-like cell line for neuronal development.
Results: In vivo, perinatal exposure to PBDE-47 induced memory deficits in adult rats. This is accompanied by hippocampal neuronal loss partly as a result of apoptosis, as evidenced by caspase-3 activation and PARP cleavage. Further study identified that PBDE-47 triggered autophagic vesicles accumulation, increased levels of microtubule-associated protein 1 light chain 3 (LC3)-II, an essential protein for autophagosomes formation, and autophagy substrate sequestosome 1 (SQSTM1/p62), but reduced levels of autophagy-related protein (ATG) 7, a key protein for autophagosomes elongation, suggestive of autophagy impairment. These findings were further demonstrated by an in vitro model of PBDE-47-treated PC12 cells. Mechanistically, autophagy alteration is more sensitive to PBDE-47 treatment than apoptosis induction. Importantly, while stimulation of autophagy by the chemical inducer rapamycin and adenovirus-mediated Atg7 overexpression aggravated PBDE-47-induced apoptosis and cell death, inhibition of autophagy by the chemical inhibitor wortmannin and siRNA knockdown of Atg7 reversed PBDE-47-produced detrimental outcomes. Interestingly, blockage of apoptosis by caspase-3 inhibitor Ac-DEVD-CHO ameliorated PBDE-47-exerted autophagy impairment and cell death, though in combination with autophagy inhibitor did not further promote cell survival.
Conclusion: Our data suggest that autophagy impairment facilitates apoptosis, which, in turn, disrupts autophagy, ultimately resulting in cell death, and that autophagy may act as a promising therapeutic target for PBDE-47-induced developmental neurotoxicity.
Keywords: PBDE-47, developmental neurotoxicity, apoptosis, autophagy, relationship
Introduction
As a major class of brominated flame retardants, polybrominated diphenyl ethers (PBDEs) have been extensively used as additives in many commodities, such as electrical equipment, textiles, building material and others. PBDEs are not chemically bound to the polymer and can readily divulge to the surrounding environment. Due to their bioaccumulation and persistence properties, PBDEs exist universally in the natural environment and biological samples [1, 2]. Despite dust inhalation and dietary intake are the primary routes of PBDEs exposure for the general population, special exposure pathways, such as transplacental transfer, breast-feeding, hand-to-mouth transfer and frequent ground contact behavior contribute to the higher body burden of PBDEs in infants and children [3]. PBDEs exposure during the critical period of brain development has attracted global attention for their developmental neurotoxicity. Mounting studies have reported that PBDEs are associated with the neurodevelopmental disorders of children and cause enduring neurobehavioral abnormalities in experimental animals after prenatal or/and postnatal exposure [4, 5]. However, the underlying mechanisms of PBDEs-induced developmental neurotoxicity are still not completely understood.
Autophagy is a lysosome-mediated dynamic self-digestion process responsible for damaged organelles and unnecessary long-lived proteins degradation and recycling [6]. The whole process of autophagy is composed of several sequential stages: initiation, elongation, maturation, fusion, degradation, and any stage is disturbed would result in autophagy dysfunction [7]. Physiologically, autophagy plays a vital role in maintaining cellular homeostasis and is indispensable for normal cellular differentiation and survival against cellular stress. However, defective autophagy has been linked to pathophysiology of various diseases such as neurodegenerative disorders and involved in cell dysfunction and death [8, 9]. Especially, the interplay of autophagy and apoptosis is intimate and intricate when determining cellular fate [10]. Autophagy is a double-edged sword that can either protect cells against apoptosis or promote apoptosis [11]. It is well documented that apoptosis is involved in PBDEs-induced developmental neurotoxicity [12-17]. Moreover, a recent study from our lab has found that acute exposure to 2,2',4,4'- tetrabromodiphenyl ether (PBDE-47), the most abundant congener in environmental and human samples, induced defective autophagy contributing to Human neuroblastoma SH-SY5Y cells death in vitro [18]. Nevertheless, little is known about how developmental PBDEs exposure would affect neuronal autophagy status. Yet the relationship between autophagy and apoptosis in PBDEs neurotoxicity has not been revealed.
In this study, we aimed to explore the role of autophagy in PBDE-47-induced developmental neurotoxicity, and particularly focus on its relationship with apoptosis in this process. Using in vivo rat model of low-dose PBDE-47 exposure from pre-pregnancy until lactation and in vitro model of PBDE-47-treated PC12 cells, a widely used neuron-like cell line for neuronal development that has been used to characterize essential features of the developmental neurotoxicity of diverse compounds [19], we found that autophagy impairment promotes excessive apoptosis, resulting in elevated cell death, and that autophagy may act as a potential therapeutic target for PBDE-47-induced developmental neurotoxicity.
Materials and Methods
Chemicals and reagents
The following antibodies were used: anti-PARP (Cell Signaling Technology, 9542), anti-caspase-3 (9661, Cell Signaling Technology, USA), anti-autophagy-related protein 7 (ATG7) (ab133528, Abcam, USA), anti-LC3 (14600-1-AP, Proteintech, USA), anti-p62 (ab56416, Abcam, USA), anti-GAPDH (60004-1-Ig, Proteintech, USA). The following chemical regents were used: PBDE-47 (purity 99.5%, GC/MS) (BDE-047N-3G, AccuStandard Corp, USA), Wortmannin (WM) (S2758, Selleck Chemicals, USA), Rapamycin (RAP) (R5000, Shanghai Haoran, China), Ac-DEVD-CHO (DEVD) (C1206-10 mM, Beyotime Institute of Biotechnology, China). All other chemical regents were analytical grade purchased from credible supplier or as described in the relevant methods.
Animals and treatments
Adult Sprague-Dawley (SD) rats purchased from the Experimental Animal Research Center of Hubei provincial Center for Disease Control and Prevention (certificate No SCXK 2015-0018, Grade SPF) were kept in the animal house maintained at temperature (22-26 °C), humidity (50%-60%), and under 12 h light/12 h dark cycle. All rats, in plastic cages, were given free access to standardized granular food and tap water. The animal experimental protocol was conducted in strict compliance with guidelines for animal care and approved by the Institutional Animal Care and Use Committee of Huazhong University of Science and Technology.
After acclimation, female rats were allocated to four groups mating with male rats at 2:1, randomly. Beginning 10 days prior to mating and ending at weaning of offspring on postnatal day (PND) 21, except the day of parturition, female rats were weighed and exposed to 0.1, 1.0, 10 mg/kg/day (1 mL/200 g body weight/day) PBDE-47 or corn oil (as vehicle control) via gavage between 9:00 and 10:00 A.M. daily. The PBDE-47 solution was obtained by dissolving the compound in corn oil and ultrasonic treatment for 30 min at room temperature (RT). The doses we chose were based on the no-observed-adverse-effect level (NOAEL, 0.7 mg/kg) and the lowest-observed-adverse-effect-level (LOAEL, 10.5 mg/kg) for developmental neurotoxicity of PBDE-47 [20], as well as our previous research that a single oral doses of PBDE-47 (1, 5, 10 mg/kg) on PND 10 impairs the learning and memory abilities in 2-month-old rats [21]. The doses are well within or above the reported range for human exposure [22] and comparable to the previous studies following developmental low-dose PBDE-47 exposure [23, 24]. Pregnant dams were raised individually in separate cages. We culled to 8 pups per litter, 4 of each sex on PND 3. The offspring were re-caged according to sex and exposure condition after weaning and kept until PND 88 (Fig. 1A). After behavioral tests, all rats were sacrificed within 24 h. The brains of female offspring rats were immediately isolated and weighted. Three brains of each group were fixed in paraformaldehyde for histopathological examination, immunohistochemical analysis, randomly. The hippocampi stripped from another three brains of each group were cut into 1 mm3 block and fixed with 2.5% glutaraldehyde for ultrastructure observation by transmission electron microscopy (TEM). The other hippocampi were frozen immediately in liquid nitrogen for 30 s and stored at -80 °C for subsequent protein extraction.
Perinatal low-dose PBDE-47 exposure induces memory impairments in adult rats. Maternal rats were treated with vehicle (corn oil) or PBDE-47 (0.1, 1.0, 10 mg/kg/day) from pre-pregnancy until weaning and female offspring rats were raised until PND 88. (A) Schedule of PBDE-47 exposure. (B) Representative traces on the fourth day in the PNT. (C) The mean escape latency, swimming distance, and swimming speed to platform in the PNT. (D) Representative traces in the SPT. (E) Distance, time spent in the target quadrant (%) and the number of platform crossings in the SPT. The data are presented for six rats each group. *P < 0.05 versus the control group.
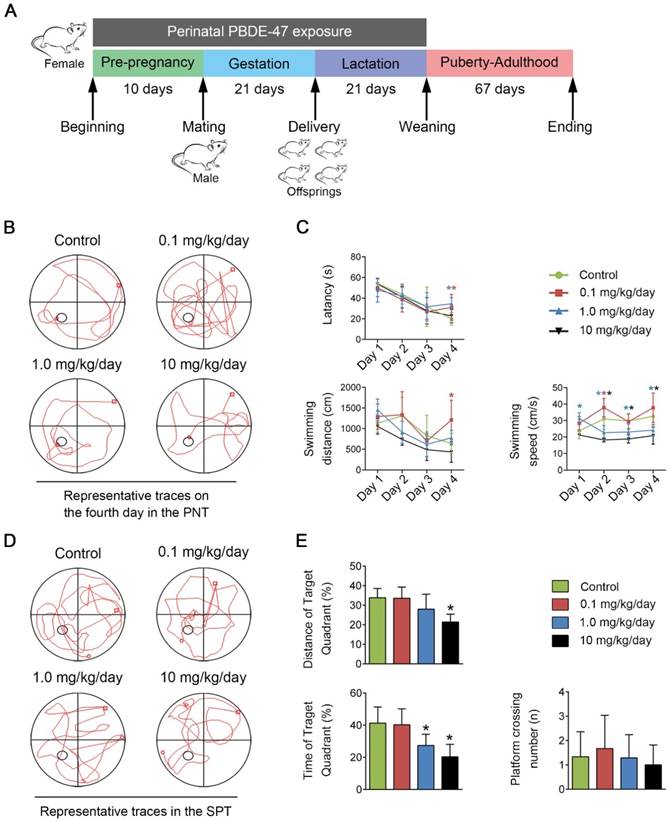
As reported recently [25], there were no signs of dysfunction, nor were there any significant difference in maternal body weight gain, relative organ weight for brain, vitality or growth of offspring rats among treatment groups in the experimental period.
Morris water maze (MWM) test
The MWM task is widely used to assess spatial learning and memory. It is consist of a black painted circular pool (1.8 m in diameter and 0.5 m high), an invisible black platform (round, 8 cm in diameter), a vidicon above the center of the pool to capture the images of swimming rats and connected to a tracking system (Electric factory of Wuhan, China). The pool was filled with water mixed with black non-toxic ink (23 ± 1 °C). Then the platform was invisible (2 cm below the liquid surface) in the center of the southwest quadrant (as the target quadrant).
The pups of several litters born within a day (one male and one female pup per litter) were selected for the MWM test. In the place navigation test (PNT), the rats were trained for four consecutive days, starting at four different quadrants of each training. The rats, facing the pool sidewall, were put into the water and given a maximum of 60 s to find the hidden platform. Those rats failed to find the platform in 60 s was guided to the platform. Both the rats found the platform by themselves and those guided by the experimenter were allowed to stay on the platform for 15 s. Then rats were toweled dry and placed in a clean cage resting for the next quadrant training. The time that rats spent finding the hidden platform was measured and showed as latency. The swimming distance, latency, and swimming speed to platform were recorded to evaluate the spatial learning ability. On the 5th day, the spatial probe test (SPT) was performed. The hidden platform was removed, and rats were put into the pool from the northeast quadrant for 60 s. The time and distance spent in the target quadrant, the number of platform crossing were recorded to measure their memory.
Histological evaluation
Nissl staining was performed according to standard laboratory procedures. Briefly, paraffin-embedded sections (4 μm) were deparaffinized, hydrated, and rinsed in distilled water for 1 min. Then the sections were stained with preheating toluidine blue solution for 30 min, rinsed quickly in distilled water for three times and differentiated in 95% ethyl alcohol until the cells were clear enough for light microscopic (Olympus, Japan) exam. Five sections were analyzed for each sample. The number of Nissl-stained neurons was measured using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, USA).
For immunohistochemistry, after being deparaffinized and rehydrated, sections were boiled in citrate buffer (10 mmol/L, pH 6.0) for antigen retrieval and incubated with 3% hydrogen peroxide for 15 min to quench endogenous peroxidase activity, subsequently blocked with phosphate-buffered saline (PBS) containing 10% (v/v) normal goat serum for 30 min. The specific primary antibody such as active caspase-3 (1:500), LC3 (1:400), and p62 (1:400) were incubated overnight at 4 °C. After being washed thrice with PBS, sections were incubated with corresponding horse-radish peroxidase (HRP)-conjugated secondary antibody (1:1000) for 1-2 h at RT. The stained sections were observed after reacting with DAB (Boster Biological Technology, AR1022) for 5 min under light microscope (Olympus, Japan). Cells showing dark brown were regarded as positive cells. Five sections were analyzed for each specimen. The positive staining were quantified using Image-Pro Plus 6.0 software.
Cell culture and treatment
PC12 (TCR 9) cells were obtained from the Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences. The cells were cultured in RPM1 1640 medium (HyClone, AD17321268) supplemented with 10% fetal bovine serum (Gibco, 1891605) at 37 °C in a humidified 5% CO2 bioclean atmosphere. PBDE-47 stock solution was prepared by dissolving PBDE-47 powder into dimethyl sulfoxide (DMSO) (Sigma-Aldrich, D8418). PBDE-47 stock solution was diluted with RPM1 1640 medium for the required concentration. To investigate the underlying mechanisms of PBDE-47 neurotoxicity, cells in exponential phase were treated with 1.0, 10, 20 μmol/L PBDE-47 for 24 h and 20 μmol/L PBDE-47 for 3, 6, 12, 24 h, respectively (DMSO as vehicle control).
In addition, to explore the relationship between apoptosis and defective autophagy, classical chemical intervention agent, such as RAP, WM, and DEVD were employed. Both RAP and WM were prepared in DMSO at a stock of 20 mmol/L and 10 mmol/L, respectively. PC12 cells were treated with PBDE-47 (20 μmol/L) with/without RAP (50 nmol/L, pre-treated for 24 h), WM (100 nmol/L) and DEVD (10 μmol/L) for 24 h. Moreover, adenovirus vector (Vigene Biosciences, CP0001) and siRNA (RiboBio, siB170712075123) mediated Atg7 overexpression and silence were implemented as specific methods to illuminate the roles of autophagy. The best-performing Atg7 siRNA sequence was GCCTCTGTATGAATTTGAA. Each cell experiment was replicated at least three times independently.
Cell viability assay
Cell viability was assessed by cell counting kit-8 assay (CCK-8; Promoter Biotechnology Ltd, B-CCK8). In detail, PC12 cells were seeded into 96-well plate (5000 cells/well) and exposed to different treatment following the cell treatment protocols. After treatment, 20 μL CCK-8 reaction mixture (mixture between CCK-8 stock solution and RPM1 1640 medium in a ratio of 1:1) was added into each well. Then, the plates were incubated at 37 °C for an additional 45 min. The cell viability was determined by optical density (OD) at 450 nm using a microplate reader (SpectraMax 190, USA).
Transmission electron microscopy (TEM)
The autophagosomal structures in hippocampal tissue and PC12 cells were visualized using TEM analysis. Hippocampal tissue pieces of 1 mm3 were immersed in 2.5% glutara ldehyde immediately after being isolated from the brain at 4 °C for 6 h. Then the pieces were fixed with 1% osmium tetroxide and dehydrated in graded ethanol series, and embedded in Araldite. Ultrathin sections (50 nm) were stained with 4% uranyl acetate for 20 min and 0.5% lead citrate for 10 min. Finally, the sections were observed under a TEM (Philips Tecnai 10). Three rats were selected randomly in each group for ultrastructure observation.
For PC12 cells, the treated cells were collected by centrifugation at 2500 rpm for 5 min. Cell aggregates were immersed in 2.5% glutaraldehyde and the latter experimental procedures were the same as the above description. Three experiments were performed independently.
Assessment of apoptosis
The apoptotic cells were determined by Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, 560931) following the manufacturer's instructions. Briefly, PC12 cells (1 × 105 cells/well) were seeded into 6-well plate and treated based on the protocols. After treatment, cells were trypsinized (no phenol red), centrifuged at 2000 rpm for 5 min. The cell pellet was resuspended with 350 μL 1 × binding buffer and stained with 5 μL Annexin V-FITC for 10 min. Ten min before being examined by the Flow Cytometer (BD Bioscience, Mountain View, CA), the cells were re-stained with 5 μL PI. All the stainings were performed at RT in the dark.
Besides, Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) assay was also performed to evaluate the apoptotic nuclear DNA breaks in PC12 cells with the In Situ Cell Death Detection Kit (Roche Applied Science, 11684795910). PC12 cells (3 × 104 cells/well) were seeded on sterile coverslips in polylysine-coated 24-well plate. After treatment, PC12 cells were fixed with cold 4% paraformaldehyde for 1 h, rinsed with PBS for thrice, permeabilized with 0.1% Triton X-100 for 5 min on ice. Then cells were rinsed with PBS for twice and incubated with fresh TUNEL reagent (prepared according to the specification) at 37 °C in the dark for 1 h. After counterstaining with DAPI staining solution (Thermo Scientific, 62248), the coverslips were moved out and mounted with anti-fluorescence quenching agent. Cells exhibiting bright green nuclear staining under laser scanning confocal microscopy (Olympus, Japan) were considered positive for apoptosis. Three sections were analyzed for each sample. Three experiments were performed independently. The TUNEL-positive cell (%) was quantified with Image-Pro Plus 6.0 software.
Western blotting analysis
The hippocampal tissues and PC12 cells were homogenized in RIPA buffer (Beyotime Institute of Biotechnology, P0013B) for 1 h and cell lysis buffer (Beyotime Institute of Biotechnology, P0013) for 0.5 h at 4 °C, respectively. Then the homogenate was centrifuged at 12,000 rpm for 15 min at 4 °C and the supernatant was collected. BCA Protein Assay Kit (Beyotime Institute of Biotechnology, P0012) was employed for the measure of the total protein concentrations. Thirty micrograms protein extracts were resolved in 10-12% SDS-polyacrylamide gels, and then transferred onto polyvinylidene fluoride membranes (Roche Applied Science, 3010040001). Firstly, the membranes were blocked with 5% (w/v) non-fat milk for 1.5 h at RT and tris-buffered saline containing 0.1% Tween-20 (TBST) acted as the solvent. Then the primary antibodies against cleaved PARP, active caspase-3, ATG7, p62 were incubated with the membranes overnight at 4 °C. GAPDH was considered as the loading control. After being washed thrice with TBST, the membranes were incubated with the corresponding HRP-conjugated secondary antibody (1:4000) (Promoter Biotechnology Ltd, P-R-HRP) for 1 h at RT. The signals were detected by ECL reagents (Advansta, 180129-22) and the bands were scanned by a gel imaging analysis system (GeneSnap version 7.05.02, SynGene, Cambridge, UK). The band density was analyzed with Quantity One 4.6.2 software (Bio-Rad, USA). Six rats were randomly selected from each group and three independent experiments were performed.
Immunofluorescence colocalization
The PC12 cells (3 × 104 cells/well) were seeded on coverslips in polylysine-coated 24-well plate. After treatment, PC12 cells were fixed with cold 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 10% goat serum for 30 min at RT, respectively. Then cells were incubated with rabbit polyclonal LC3 antibodies (1:500) overnight at 4 °C. After thrice rinse, the following protocols were performed in the dark: Alexa Fluor 594 AffiniPure goat anti-Rabbit IgG (H+L) (1:400) (Promoter Biotechnology Ltd, P-R-594) acted as the secondary antibody was incubated for 1.5 h at RT. Whereafter, cells were counterstained with TUNEL reaction mixture at 37 °C for 1 h and the cell nuclear was stained with DAPI staining solution at 37 °C for 15 min. After mounting, fluorescent images were captured by laser scanning confocal microscopy (Olympus, Japan). LC3-II-positive puncta was also quantified with Image-Pro Plus 6.0 software.
Statistical analysis
Data were presented as mean ± S.D., and differences among the mean in each group were tested by one-way ANOVA followed by a Student-Newman-Keuls test with SPSS 21.0 (IBM SPSS, Inc., USA). P < 0.05 was considered significant.
Results
Perinatal low-dose PBDE-47 exposure induces memory impairments in adult rats
To evaluate the learning and memory abilities of adult offspring rats after perinatal low-dose PBDE-47 exposure, the MWM test was performed. For the PNT of MWM task, compared with the control rats, 0.1 mg/kg/day PBDE-47-treated rats had worse performance, whose latency and swimming distance were significantly increased on the 4th day (P < 0.05); in addition, the longer latency of 1.0 mg/kg/day PBDE-47-treated group were also observed on the 4th day (P < 0.05); however, the swimming speed of 1.0 and 10 mg/kg/day PBDE-47-treated rats were significantly decreased starting from the 2nd day (P < 0.05) (Fig. 1B-C). For the SPT of MWM task, 1.0 and 10 mg/kg/day PBDE-47-treated rats showed reduced swimming time in the target quadrant than the control rats, with an additional significant decreased swimming distance in the target quadrant of 10 mg/kg/day PBDE-47 treated rats (Fig. 1D-E). We found no significant statistical interaction between perinatal PBDE-47 exposure and sex in behavioral test (data not shown). Together, these results indicate that perinatal PBDE-47 exposure at the two higher doses induces memory deficits in adult rats.
Perinatal low-dose PBDE-47 exposure induces hippocampal neuronal loss accompanied by excessive apoptosis and defective autophagy in adult rats
Hippocampus is a vital region required for mammalian learning and memory. We used Nissl staining to evaluate the number of hippocampal neurons. As shown in Fig. 2A, the control hippocampal neurons had abundant Nissl bodies and were arranged regularly. In contrast, PBDE-47-treated rats showed fewer Nissl-stained neurons specifically in the hippocampal CA3 region, and the cells had irregular morphology with a disordered arrangement, especially in 1.0 and 10 mg/kg/day groups. The results indicate that perinatal PBDE-47 exposure results in the neuronal loss in hippocampal CA3 region of adult rats.
Perinatal low-dose PBDE-47 exposure induces hippocampal neuronal loss accompanied by excessive apoptosis and defective autophagy in adult rats. Maternal rats were exposed to vehicle (corn oil) or PBDE-47 (0.1, 1.0, 10 mg/kg/day) by oral gavage daily during the perigestational period and female offspring rats were raised until PND 88. (A) Representative images of Nissl and immunohistochemical staining and the corresponding quantifications in hippocampal CA3 region of female rats. The first row: scale bar represents 500 μm; other rows: scale bar represents 50 μm. Black arrows: positive-staining neurons (brown). (B) Representative western blotting and quantifications of apoptosis markers cleaved PARP and active caspase-3 in hippocampal tissues of female rats. (C) Ultrastructural changes in hippocampal CA3 region of the female rats. Top row: scale bar represents 2 μm; bottom row: scale bar represents 500 nm. Blue arrows: autophagosome vesicles. (D) Representative western blotting of autophagy markers ATG7, LC3-II, p62 in hippocampal tissues of female rats. (E) Quantitative analysis of autophagy markers. The data are presented as the mean ± S.D. *P < 0.05 versus the control group.
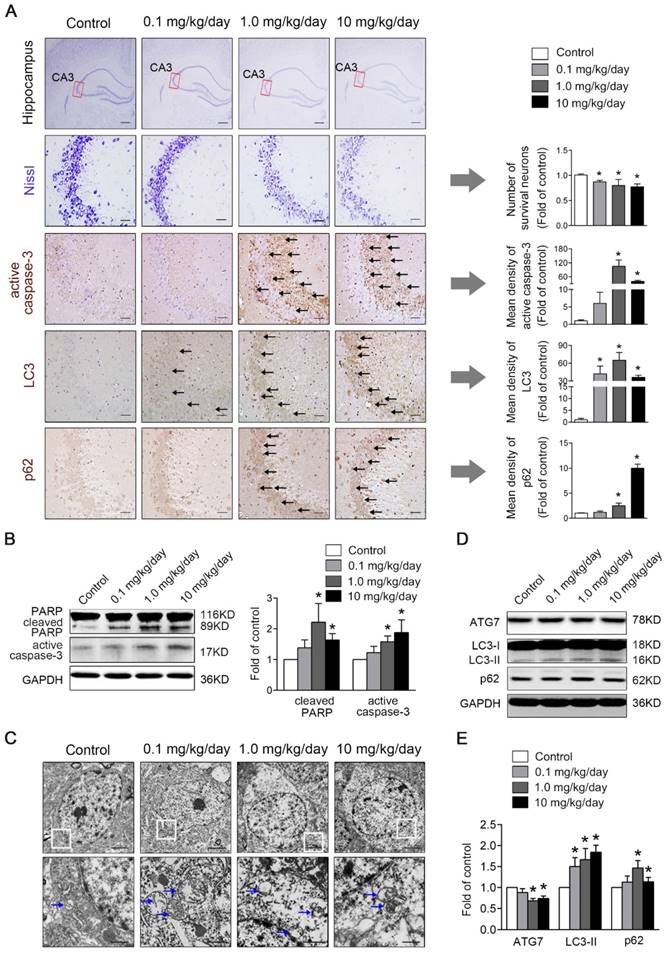
As apoptosis plays a vital role in the regulation of cell number, we investigated whether apoptosis was involved in PBDE-47-induced hippocampal neuronal loss. As shown in Fig. 2B, western blotting analysis revealed that the levels of apoptosis marker proteins active caspase-3 and cleaved PARP, were dramatically increased in 1.0 and 10 mg/kg/day PBDE-47-treated group compared to the control group (P < 0.05). Consistently, immunohistochemical staining also showed an enhanced cytoplasmic staining for active caspase-3 in 1.0 and 10 mg/kg/day PBDE-47-treated group (Fig. 2A), implying that perinatal exposure to PBDE-47 at the two higher doses induces hippocampal neuronal apoptosis in adult rats.
To determine the involvement of autophagy in PBDE-47-induced hippocampal neuronal dysfunction, TEM, the gold standard method for detection of autophagy, was employed to verify the presence of autophagosomes in hippocampal neurons. As shown in Fig. 2C, increased autophagosome-like double-membrane vesicles were observed in PBDE-47-treated groups when compared with the control group. To confirm the increased autophagosome vesicles observed by TEM, we detected the levels of microtubule-associated protein 1 light chain 3 (MAP1LC3B/LC3), the most widely used autophagic marker protein. LC3-II is localized on both the inner and outer membranes of autophagosome, the amount of which is correlated with the number of autophagosomes [26]. Western blotting analysis revealed that the levels of LC3-II were significantly increased in hippocampal neurons of PBDE-47 treated rats (Fig. 2D-E).
Moreover, the LC3 staining was notably increased in PBDE-47-exposed hippocampal neurons by immunohistochemical staining (Fig. 2A), confirming the autophagosomes accumulation. To investigate whether autophagy is induced by PBDE-47 exposure, we then examined the process of autophagy by analyzing the autophagy marker proteins including ATG7 and p62. As shown in Fig. 2D-E, the levels of ATG7, a key protein responsible for the expansion of autophagosomes, were dramatically decreased in 1.0 and 10 mg/kg/day PBDE-47-treated groups, indicating the suppression of autophagosomes formation. In contrast, the levels of p62, a substrate of selective autophagy that is accumulated under autophagy inhibition, were increased in the same doses (Fig. 2D-E), accompanied by the increased p62 staining in hippocampal neurons in comparison to controls (Fig. 2A), suggestive of blockage of autophagic degradation. Overall, these findings suggest that perinatal PBDE-47 exposure inhibits the autophagosomes formation and blocks the autophagic degradation, resulting in autophagosomes accumulation and inducing autophagy impairment in hippocampal neurons of adult rats.
PBDE-47-induced autophagy alteration is more sensitive than apoptosis induction in PC12 cells
We next asked whether the above-observed changes could occur in PC12 cells in vitro. Firstly, the effects of PBDE-47 on PC12 cells apoptosis were determined. As shown in Fig. 3A, the percentages of early and total apoptotic cells were significantly increased in 20 μmol/L PBDE-47-treated group, with an additional significant increase in the percentage of total apoptotic cells in 10 μmol/L PBDE-47-treated group (P < 0.05). Subsequently, we also used the TUNEL assay to assess the apoptosis-related DNA fragmentation. As shown in Fig. 3B, treatment with 20 μmol/L PBDE-47 notably increased the number of TUNEL-positive cells (%). Consistent with these, western blotting analysis demonstrated that 10 μmol/L and 20 μmol/L PBDE-47 treatment dramatically increased the protein levels of active caspase-3 and cleaved PARP (P < 0.05) (Fig. 3C-D). Taken together, these results suggest that PBDE-47 treatment induces excessive apoptosis of PC12 cells in a dose-dependent manner.
PBDE-47-induced autophagy alteration is more sensitive than apoptosis induction in PC12 cells. PC12 cells were exposed to 1.0, 10, 20 μmol/L PBDE-47 for 24 h and treated with 20 μmol/L PBDE-47 for 3 h, 6 h, 12 h, 24 h (DMSO as the vehicle control). (A) Flow cytometry analysis for the percentage of apoptotic PC12 cells after PBDE-47 exposure. (B) TUNEL staining of PC12 cells. Green: TUNEL-positive staining; blue: DAPI staining. Scale bar represents 10 μm. (C) Representative western blotting for apoptosis markers in PC12 cells. (D) Quantitative analysis of apoptosis markers. (E) Representative electron micrographs of PC12 cells after PBDE-47 exposure. Top row: scale bar represents 2 μm; bottom row: scale bar represents 500 nm. Blue arrows: autophagosomes. (F) Representative confocal micrographs and quantification of LC3-positive puncta in PC12 cells. Red: LC3-positive puncta; blue: DAPI staining. Scale bar represents 10 μm. (G) Representative western blotting for autophagy markers. (H) Quantitative analysis of autophagy markers. (I) Representative western blotting for active caspase-3 and LC3-II at different time points. (J) Quantitative analysis of active caspase-3 and LC3-II at different time points. The data are presented as the mean ± S.D. of three independent experiments. *P < 0.05 versus the control group.
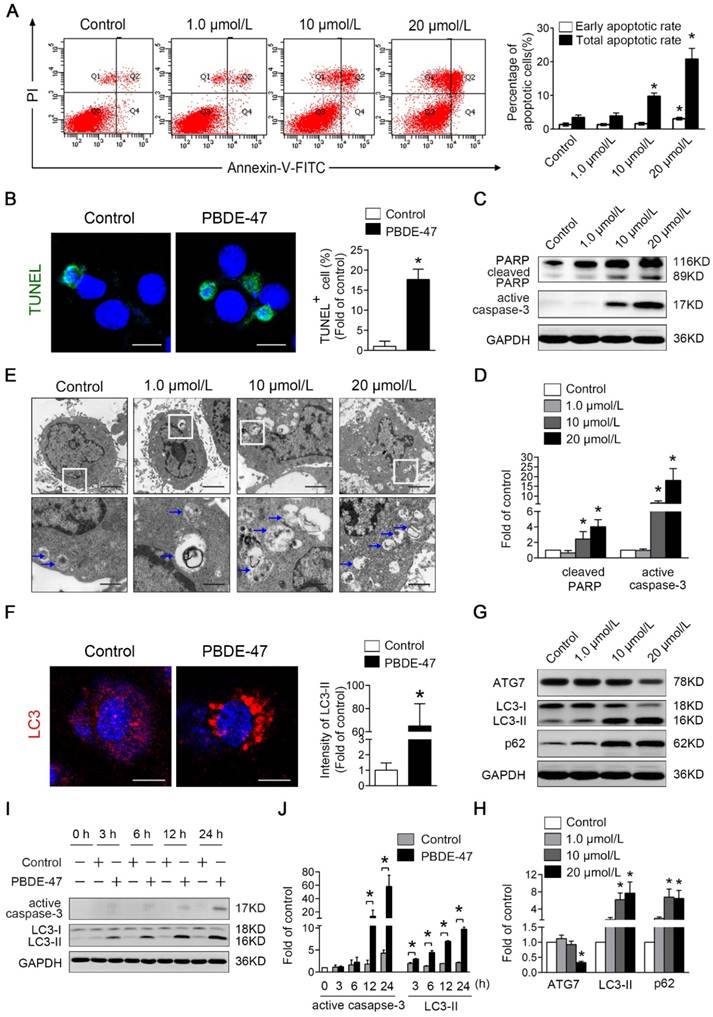
Secondly, the influences of PBDE-47 on PC12 cells autophagy activity were evaluated. As shown in Fig. 3E, TEM demonstrated that the increased number of typical double-membrane autophagosomes was visualized upon PBDE-47 treatment compared with the control group. In support of this, confocal microscopy revealed that PBDE-47 induces the accumulation of LC3-positive puncta in PC12 cells (Fig. 3F). Moreover, the level of ATG7 was significantly decreased in 20 μmol/L PBDE-47 group, while the levels of LC3-II and p62 were notably increased in 10 μmol/L and 20 μmol/L PBDE-47 groups (P < 0.05) (Fig. 3G-H). These data strongly suggest that PBDE-47 treatment results in defective autophagy with autophagosomes accumulation in PC12 cells via inhibition of autophagosomes formation and autophagic degradation.
Next, the time effects of PBDE-47 on apoptosis and autophagy changes were also determined throughout 24 h. As displayed in Fig. 3I-J, the protein levels of LC3-II were significantly increased starting from 3 h and this increase was sustained up to 24 h, while the levels of active caspase-3 were elevated dramatically starting from 12 h until 24 h after PBDE-47 treatment (P < 0.05). Taken together, these results indicate that PBDE-47-induced autophagy alteration is more sensitive than apoptosis induction in PC12 cells.
Stimulation of autophagy aggravates PBDE-47-induced apoptosis promoting PC12 cells death
Given that the autophagy alteration is more sensitive than apoptosis induction and the autophagosome formation is blocked following PBDE-47 exposure, we first sought to explore the effect of autophagy promotion on PBDE-47-induced apoptosis. As shown in Fig. 4A, pharmacological activation of autophagy with RAP, a specific mTOR inhibitor known to stimulate autophagy, increased the level of LC3-II, which further increased the levels of active caspase-3 and cleaved PARP, thus aggravating apoptosis induced by PBDE-47. Moreover, genetic stimulation of autophagy by infection of adenovirus expressing Atg7 (Ad-Atg7) significantly increased cleaved PARP, while decreased the level of active caspase-3 when compared with PBDE-47 treatment alone (P < 0.05) (Fig. 4B). Given the opposite changes in cleaved PARP and active caspase-3, caspase-3 inhibitor DEVD was used to clarify the role of apoptosis in this context. As shown in Fig. 4C, the levels of active caspase-3 and especially cleaved PARP were dramatically decreased after DEVD administration, in comparison to PBDE-47 treatment alone or in combination with Ad-Atg7 infection (both P < 0.05). In addition, we also performed the immunofluorescence colocalization of LC3-positive puncta with TUNEL-positive cells to further confirm the effect of autophagy stimulation by Atg7 overexpression on PBDE-47-induced apoptosis.
Stimulation of autophagy aggravates PBDE-47-induced apoptosis promoting PC12 cells death. PC12 cells were treated with 20 μmol/L PBDE-47 for 24 h with/without autophagy inducer RAP (50 nmol/L) and Ad-Atg7 infection (MOI=200) (pre-treated for 24 h). Cells infected with control adenovirus (Ad-Null) were treated as control. (A) Representative western blotting and quantifications of autophagy and apoptosis markers in PC12 cells after PBDE-47 combined with RAP treatment. (B) Representative western blotting and quantifications of autophagy and apoptosis markers in PC12 cells after combination of Ad-Atg7 infection and PBDE-47 treatment. (C) Representative western blotting and quantifications of apoptosis markers in PC12 cells after combination of Ad-Atg7 infection with caspase-3 inhibitor DEVD. (D) Representative confocal micrographs of immunofluorescence for LC3-positive puncta and TUNEL-positive PC12 cells after Ad-Atg7 infection. Green: TUNEL-positive staining; red: LC3-positive puncta; blue: DAPI staining. Scale bar represents 10 μm. (E) CCK-8 analysis of cell viability after combination of PBDE-47 and RAP treatment. (F) CCK-8 analysis of cell viability after Ad-Atg7 infection plus PBDE-47 treatment. The data are presented as the mean ± S.D. of three independent experiments. *P < 0.05 versus the control group. #P < 0.05 versus the PBDE-47 group. &P < 0.05 versus the combination of Ad-Atg7 infection and PBDE-47 treatment group.
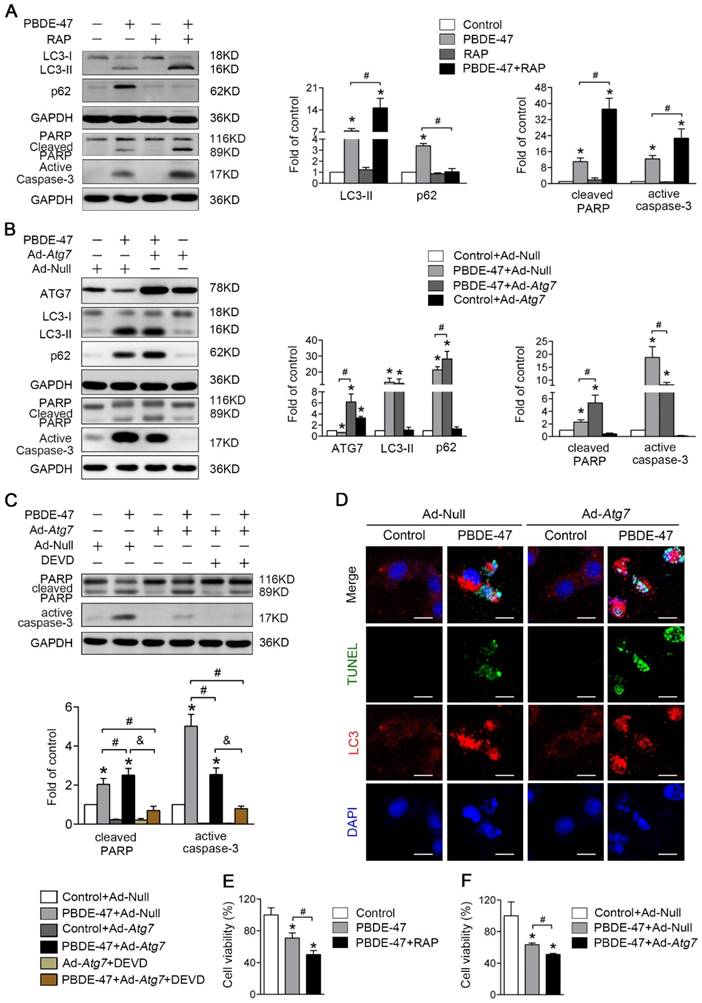
As shown in Fig. 4D, Atg7 overexpression further increased the number of TUNEL-positive cells containing LC3-positive puncta (red) following PBDE-47 treatment, supporting the notion that stimulation of autophagy aggravates PBDE-47-induced apoptosis. Importantly, both pharmacologically and genetically stimulating autophagy further decreased cell viability (Fig. 4E-F). Taken together, the above findings suggest that stimulation of autophagy exacerbates apoptosis thus promoting PC12 cells death following PBDE-47 treatment.
Inhibition of autophagy attenuates PBDE-47-induced apoptosis alleviating PC12 cells death
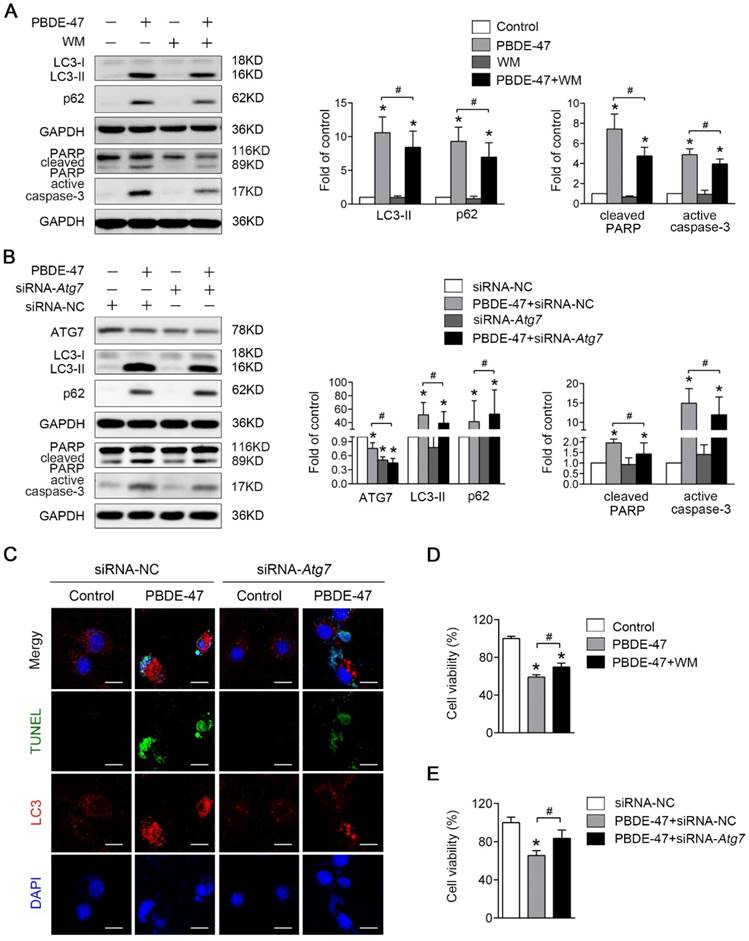
Next, we asked whether inhibition of autophagy would have the converse effect on PBDE-47-induced apoptosis. As expected, pharmacological suppression of autophagy with WM, a potent PI3-kinase inhibitor that was extensively used to inhibit autophagy induction [27], decreased the level of LC3-II, and dramatically diminished PBDE-47-induced apoptosis, evidenced by decreased protein levels of active caspase-3 and cleaved PARP (P < 0.05) (Fig. 5A). Consistently, using specific siRNAs to knockdown Atg7 further attenuate apoptosis triggered by PBDE-47 (P < 0.05) (Fig. 5B). To verify the effect of inhibiting autophagy by siRNA-Atg7 transfection, the immunofluorescence colocalization of LC3-positive puncta with TUNEL-positive cells was carried out. Not surprisingly, confocal microscopy revealed that the number of PBDE-47-induced TUNEL-positive cells with less LC3-positive puncta accumulation was notably decreased after siRNA-Atg7 transfection, supporting the idea that inhibition of autophagy alleviates PBDE-47-induced apoptosis (Fig. 5C). Furthermore, the cell viability was significantly increased after further inhibiting autophagy either pharmacologically or genetically relevant to PBDE-47 treatment alone (P < 0.05) (Fig. 5D-E). Taken together, the results indicate that suppression of autophagy attenuates apoptosis thereby reducing PC12 cells death under PBDE-47 treatment.
Inhibition of autophagy attenuates PBDE-47-induced apoptosis alleviating PC12 cells death. PC12 cells were treated with 20 μmol/L PBDE-47 for 24 h in the presence or absence of autophagy inhibitor, WM (100 nmol/L) and siRNA-Atg7 transfection (50 nmol/L). (A) Representative western blotting and quantifications of the levels of autophagy and apoptosis markers in PC12 cells after PBDE-47 plus WM treatment. (B) Representative western blotting and quantifications of autophagy and apoptosis markers in PC12 cells after combination of siRNA-Atg7 transfection and PBDE-47 treatment. (C) Representative confocal images of immunofluorescence for LC3-positive puncta and TUNEL-positive PC12 cells after siRNA-Atg7 transfection. Green: TUNEL-positive staining; red: LC3-positive puncta; blue: DAPI staining. Scale bar represents 10 μm. (D) CCK-8 analysis of cell viability after combination of PBDE-47 with WM treatment. (E) CCK-8 analysis of cell viability after transfection of siRNA-Atg7 followed by PBDE-47 treatment. The data are presented as the mean ± S.D. of three independent experiments. *P < 0.05 versus the control group. #P < 0.05 versus the PBDE-47 group.
Blockage of apoptosis mitigates PBDE-47-induced defective autophagy facilitating PC12 cells survival
We also asked whether apoptosis plays a role in autophagy impairment under such circumstance. As shown in Fig. 6A, combination of caspase-3 inhibitor DEVD and PBDE-47 treatment significantly decreased the level of LC3-II, indicative of reduced autophagosomes accumulation compared with PBDE-47 treatment alone. Correspondingly, DEVD incubation resulted in an increase in cell viability compromised by PBDE-47 treatment (P < 0.05) (Fig. 6B). These results suggest that blockage of apoptosis attenuates the autophagosomes accumulation promoting PC12 cells survival following PBDE-47 treatment.
Blockage of apoptosis mitigates PBDE-47-induced defective autophagy facilitating PC12 cells survival. PC12 cells were treated with 20 μmol/L PBDE-47 combined with 10 μmol/L DEVD for 24 h. (A) Representative western blotting and quantifications of apoptosis and autophagy markers in PC12 cells after PBDE-47 combined DEVD treatment. (B) CCK-8 analysis of cell viability after PBDE-47 co-treatment with DEVD administration in the presence or absence of WM. The data are presented as the mean ± S.D. of three independent experiments. *P < 0.05 versus the control group. #P < 0.05 versus the PBDE-47 group.
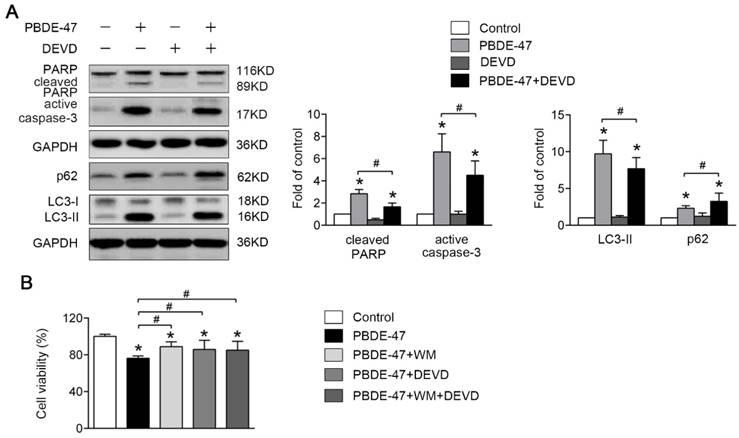
Finally, we investigated whether the defective autophagy and excessive apoptosis promote cell death with combined effect. To this end, the autophagy inhibitor WM was used in combination with the apoptosis blocker DEVD to investigate the effects of both responses on cell survival together. Interestingly, co-treatment with DEVD and WM did not further reverse PBDE-47-induced decrease in cell viability compared with either of them alone, suggesting defective autophagy and excessive apoptosis may be coupled events.
Discussion
In this study, we used an in vivo rat model of long-term of PBDE-47 exposure from pre-pregnancy until offspring weaning to mimic human exposure. We found that perinatal exposure to PBDE-47 at the two higher doses induced memory deficits in adult rats, which were based on the decreased swimming time and distance in the target quadrant, the more robust parameters for short-term memory [28, 29], and took the interference of swimming speed into account in MWM test. Our results are consistent with previous studies that perinatal exposure to PBDE-47 at comparable dosage (1.0 mg/kg/day) caused impaired performance of adult mice in the MWM and Barnes maze test [23, 30]. Interestingly, these results coincide with the epidemiological studies that exposure to PBDEs during both prenatal and postnatal periods are associated with poor memory and cognition in early adolescence [31, 32]. Our findings thus confirm and contribute to the growing evidence that PBDEs are developmental neurotoxicants.
The exact mechanisms involved in developmental neurotoxicity after PBDE-47 exposure are still indistinct and the direct effects on the developing brain are one of the frequently studied modes of action. In the present study, exposure to 1.0 and 10 mg/kg/day PBDE-47 resulted in obvious neuronal loss in hippocampal CA3 region, as evidenced by Nissl staining. Since apoptosis plays a crucial role in maintaining neuronal number [33], it is postulated that apoptosis may be involved. Indeed, we further provided adequate evidence that PBDE-47 exposure induced neuronal apoptosis, as demonstrated by increased caspase-3 activation, and PARP cleavage, which is confirmed by in vitro results with additional evidence that inhibiting apoptosis promotes PC12 cells survival. Our data consistent with the notion that PBDE-47 induces apoptosis resulting in neuronal loss [12-16, 34], and support the important role of apoptosis in developmental neurotoxicity of perinatal PBDE-47 exposure.
Autophagy serves as a protective mechanism for promoting the cell survival, while defective autophagy has been implicated in cell loss [9]. However, the studies on the roles of autophagy in PBDEs toxicity are still lacking. Limited studies reported that both PBDE-100 and PBDE-153 treatment resulted in accumulation of autophagosomes involved in HepG2 cells injury [35, 36]. In the present study, PBDE-47 triggered autophagosomes accumulation identified by the increased double-membrane autophagosome vesicles, LC3-positive puncta and levels of LC3-II both in vivo and in vitro. However, accumulated autophagosomes results from either induction of autophagosomes formation or disruption of autophagosomes degradation [37]. It is known that the level of LC3-II is directly associated with the amount of mature autophagosomes and accumulated p62 is always attributed to a block in autophagosomes degradation [26, 38]. Therefore, it is likely that the increased levels of LC3-II and p62 are attributed to the blockage of autophagic degradation. Indeed, our latest results show that PBDE-47 induces lysosomal dysfunction and disturbs fusion of lysosomes with autophagosomes, contributing to the impaired autophagic degradation (unpublished data). Meanwhile, the levels of ATG7, playing a vital role in the expansion of autophagosomes [39], were dramatically decreased, which suggests the suppression of autophagosomes formation. Taken together, it's conceivable that PBDE-47 exposure inhibits autophagosomes formation and, more importantly, blocks their degradation, thereby resulting in autophagosomes accumulation and autophagy impairment. This defective autophagy leads to cell death as verified by stimulating and inhibiting autophagy pharmacologically and genetically. Our findings are consistent with a recent study from our lab that PBDE-47 exposure led to autophagosomes accumulation contributing to SH-SY5Y cells death [18]. However, perigestational exposure to the same doses of PBDE-47 inhibited autophagy activity with reduced levels of LC3-II and increased p62 accumulation conducing to maternal thyroid cells death [25]. Interestingly, PBDE-209 exposure during pregnancy induced excessive autophagy associated with the disturbance of learning and memory abilities in the offspring rats [40]. Differences in cell-type specificity, dose sensitivity, duration of treatment and congener structure may explain the inconsistences.
Apoptosis and autophagy play pivotal roles in the determination of cell fate, while the interplay between these two processes is very complicated in this context. Given PBDE-47 caused the blockage of autophagosomes formation, stimulation of autophagy was first used to observe the impact of autophagy on apoptosis. Pharmacological induction of autophagy aggravated the autophagosomes accumulation due to failed degradation, which further enhanced PBDE-47-induced PC12 cells apoptosis manifested as caspase-3 activation and PARP cleavage. Interestingly, although genetic stimulation of autophagy by Atg7 overexpression increased PARP cleavage, the caspase-3 activation was reduced. However, using caspase-3 inhibitor DEVD, we have confirmed the role of apoptosis in this process as the levels of both active caspase-3 and cleaved PARP were dramatically decreased, when compared with PBDE-47 treatment alone or in combination with Ad-Atg7 infection. In addition, Atg7 overexpression increased the number of TUNEL-positive cells containing LC3-positive puncta following PBDE-47 treatment, further supporting the notion that stimulation of autophagy aggravates PBDE-47-induced apoptosis. Undoubtedly, the cell survival was further compromised under the circumstances. Our results are consistent with the study that induction of autophagy by RAP future potentiated PCB28- and PCB52-triggered L-02 and Brl-3A cells apoptosis and hepatotoxicity [41].
An important finding of the present study is that inhibition of autophagy attenuated apoptosis and promoted cell survival. Both pharmacological and genetic inhibition of autophagy alleviated PBDE-47-induced PC12 cells apoptosis, as firmly evidenced by the mitigation of caspase-3 activation, PARP cleavage and DNA fragmentation. This contributes to the elevated cell survival and may be due to the decreased autophagosomes accumulation since the level of LC3-II was reduced. These findings suggest that autophagy impairment, more precisely, the abnormal autophagosomes accumulation facilitates apoptosis induction under PBDE-47 treatment, which is further supported by the reduced LC3-positive puncta within decreasing TUNEL-positive cells after suppression of autophagy. Our results are in agreement with a previous study from our lab that inhibiting autophagy induction by 3-methyladenine (3-MA), another potent PI3-kinase inhibitor similar to WM, promoted SH-SY5Y cells survival following PBDE-47 exposure [18]. More importantly, inhibition of autophagy by 3-MA also diminished PBDE-209-induced apoptosis in primary hippocampal neurons [40], supporting our findings and suggesting that targeting inhibition of autophagy may be a promising therapeutic target for the prevention and treatment of PBDEs neurotoxicity. Interestingly, some other studies also showed that apoptosis was enhanced upon inhibition of autophagy with WM in HepG2 cells following PBDE-100 or PBDE-153 treatment [35, 36], perhaps due to differences in the cell-type specificity and congener structure.
Interestingly, blockage of apoptosis by inhibiting apoptotic executioner, caspase-3, also alleviated the autophagosomes accumulation, as demonstrated by a reduction in LC3-II. Conversely, a slightly increased level of p62 suggests that the inhibition of autophagic degradation still exists even if blocking apoptosis execution. Actually, caspase-3 has been found to directly interact with key ATGs implicated in the autophagic cascade. It's reported that caspase-3 activation is able to inhibit autophagy by cleaving ATG5 and ATG6 [42, 43], while induce autophagy by cleaving ATG4D [44]. Therefore, further studies are required to reveal the interplay between ATGs and caspases under such neurotoxic process. Moreover, the combination of autophagy inhibition with apoptosis blockage did not further promote cell survival following PBDE-47 treatment compared to each of them alone, suggesting that the defective autophagy and excessive apoptosis may be coupled events.
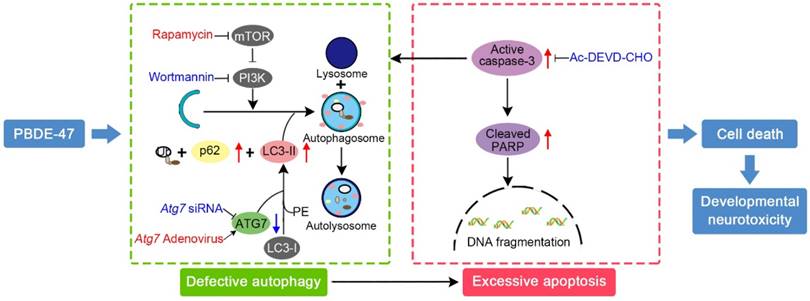
In conclusion, we provide in vivo and in vitro evidence demonstrating that the impaired autophagy, resulting from suppression of both autophagosomes formation and degradation aspects, contributes to PBDE-47-induced developmental neurotoxicity in addition to excessive apoptosis. Particularly, we have revealed that this autophagy impairment facilitates apoptosis, resulting in increased cell death (Fig. 7). These findings offer novel insights into a better understanding of the underlying mechanisms responsible for developmental PBDE-47 neurotoxicity and a theoretical support for autophagy as a promising therapeutic target for diminishing PBDE-47 neurotoxicity. However, it should be noted that additional studies are still needed to confirm our present findings using primary cultured neurons. Work is ongoing on samples from male rats perinatally exposed to the same doses of PBDE-47 at later time points with the aim of better understanding the mechanisms of action of PBDE-47 and the effects of PBDE-47 long after cease of exposure.
Schematic model illustrates the role of autophagy in PBDE-47-induced developmental neurotoxicity and its relationship with apoptosis. PBDE-47 increased levels of LC3-II, an essential protein for autophagosomes formation, and autophagy substrate p62, but reduced levels of ATG7, a key protein for the elongation of autophagosomes, suggestive of autophagy impairment. These, together with excessive apoptosis as evidenced by caspase-3 activation and PARP cleavage, contributes to PBDE-47-induced cell death in vivo and in vitro. Stimulation of autophagy by the chemical inducer rapamycin and Atg7 overexpression aggravated PBDE-47-induced apoptosis and cell death, while inhibition of autophagy by the chemical inhibitor wortmannin and siRNA knockdown of Atg7 reversed PBDE-47-produced detrimental outcomes. Blockage of apoptosis by caspase-3 inhibitor Ac-DEVD-CHO ameliorated PBDE-47-exerted autophagy impairment and cell death. As a result, PBDE-47-induced autophagy impairment facilitates apoptosis, which, in turn, disrupts autophagy, ultimately resulting in enhanced cell death and developmental neurotoxicity.
Abbreviations
ATG: autophagy-related protein; CCK-8: cell counting kit-8; DAPI: 4',6-diamidino-2-phenylindole; DEVD: Ac-DEVD-CHO; DMSO: dimethyl sulfoxide; FITC: fluorescein isothyocyanate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HRP: horse-radish peroxidase; LC3: microtubule-associated protein 1 light chain 3; LOAEL: lowest-observed-adverse-effect-level; MWM: Morris water maze; NOAEL: no-observed-adverse-effect level; PBDE-47: 2,2',4,4'- tetrabromodiphenyl ether; PBDEs: polybrominated diphenyl ethers; PBS: phosphate-buffered saline; PI: propidine iodide; PND: postnatal day; PNT: place navigation test; RAP: rapamycin; RT: room temperature; SD: Sprague-Dawley; SPF: specific pathogen free; SPT: spatial probe test; SQSTM 1/p62: sequestosome 1; TEM: transmission electron microscopy; TUNEL: terminal deoxynucleotidyl transferase mediated dUTP nick end labeling; WM: wortmannin.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grants No. 81773388 and No. 81502785), the State Key Program of National Natural Science of China (Grant No. 81430076) and the Fundamental Research Funds for the Central Universities (HUST 2016YXMS221 and HUST 2015ZDTD052).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Law RJ, Covaci A, Harrad S. et al. Levels and trends of PBDEs and HBCDs in the global environment: status at the end of 2012. Environ Int. 2014;65:147-58
2. Vuong AM, Yolton K, Dietrich KN. et al. Exposure to polybrominated diphenyl ethers (PBDEs) and child behavior: Current findings and future directions. Horm Behav. 2017;101:94-104
3. Linares V, Bellés M, Domingo JL. Human exposure to PBDE and critical evaluation of health hazards. Arch Toxicol. 2015;89:335-56
4. Lam J, Lanphear BP, Bellinger D. et al. Developmental PBDE Exposure and IQ/ADHD in Childhood: A Systematic Review and Meta-analysis. Environ Health Perspect. 2017;125:86001
5. Costa LG, de Laat R, Tagliaferri S. et al. A mechanistic view of polybrominated diphenyl ether (PBDE) developmental neurotoxicity. Toxicol Lett. 2014;230:282-94
6. Galluzzi L, Baehrecke EH, Ballabio A. et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811-36
7. Chen G, Ke Z, Xu M. et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8:1577-89
8. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805-11
9. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65-75
10. Mariño G, Niso-Santano M, Baehrecke EH. et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Bio. 2014;15:81-94
11. Ravegnini G, Sammarini G, Nannini M. et al. Gastrointestinal stromal tumors (GIST): Facing cell death between autophagy and apoptosis. Autophagy. 2017;13:452-63
12. Chen H, Tang X, Zhou B. et al. A ROS-mediated mitochondrial pathway and Nrf2 pathway activation are involved in BDE-47 induced apoptosis in Neuro-2a cells. Chemosphere. 2017;184:679-86
13. Costa LG, Pellacani C, Dao K. et al. The brominated flame retardant BDE-47 causes oxidative stress and apoptotic cell death in vitro and in vivo in mice. Neurotoxicology. 2015;48:68-76
14. He P, He W, Wang A. et al. PBDE-47-induced oxidative stress, DNA damage and apoptosis in primary cultured rat hippocampal neurons. Neurotoxicology. 2008;29:124-29
15. Zhang S, Kuang G, Zhao G. et al. Involvement of the mitochondrial p53 pathway in PBDE-47-induced SH-SY5Y cells apoptosis and its underlying activation mechanism. Food Chem Toxicol. 2013;62:699-706
16. Zhang S, Chen Y, Wu X. et al. The Pivotal Role of Ca2+ Homeostasis in PBDE-47-Induced Neuronal Apoptosis. Mol Neurobiol. 2016;53:7078-88
17. He P, Wang AG, Xia T. et al. Mechanisms underlying the developmental neurotoxic effect of PBDE-47 and the enhanced toxicity associated with its combination with PCB153 in rats. Neurotoxicology. 2009;30:1088-95
18. Zhang C, Li P, Zhang S. et al. Oxidative stress-elicited autophagosome accumulation contributes to human neuroblastoma SH-SY5Y cell death induced by PBDE-47. Environ Toxicol Pharmacol. 2017;56:322-28
19. Slotkin TA, MacKillop EA, Ryde IT. et al. Ameliorating the developmental neurotoxicity of chlorpyrifos: a mechanisms-based approach in PC12 cells. Environ Health Perspect. 2007;115:1306-13
20. U.S. Environmental Protection Agency: Washing, DC. Toxicological Review of 2,2',4,4'-tetrabromodiphenyl Ether (BDE-47) in Support of Summary Information on the Integrated Risk Information System (IRIS). June 2008. https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P1000PLX.txt
21. He P, Wang A, Niu Q. et al. Toxic effect of PBDE-47 on thyroid development, learning, and memory, and the interaction between PBDE-47 and PCB153 that enhances toxicity in rats. Toxicol Ind Health. 2011;27:279-88
22. Johnson-Restrepo B, Kannan K, Rapaport DP. et al. Polybrominated diphenyl ethers and polychlorinated biphenyls in human adipose tissue from New York. Environ Sci Technol. 2005;39:5177-82
23. Ta TA, Koenig CM, Golub MS. et al. Bioaccumulation and behavioral effects of 2,2',4,4'-tetrabromodiphenyl ether (BDE-47) in perinatally exposed mice. Neurotoxicol Teratol. 2011;33:393-404
24. Talsness CE, Kuriyama SN, Sterner-Kock A. et al. In utero and lactational exposures to low doses of polybrominated diphenyl ether-47 alter the reproductive system and thyroid gland of female rat offspring. Environ Health Perspect. 2008;116:308-14
25. Li P, Liu L, Zhou G. et al. Perigestational exposure to low doses of PBDE-47 induces excessive ER stress, defective autophagy and the resultant apoptosis contributing to maternal thyroid toxicity. Sci Total Environ. 2018;645:363-71
26. Kabeya Y, Mizushima N, Ueno T. et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720-28
27. Vakifahmetoglu-Norberg H, Xia HG, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015;125:5-13
28. Remondes M, Schuman EM. Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature. 2004;431:699-703
29. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848-58
30. Koenig CM, Lango J, Pessah IN. et al. Maternal transfer of BDE-47 to offspring and neurobehavioral development in C57BL/6J mice. Neurotoxicol Teratol. 2012;34:571-80
31. Eskenazi B, Chevrier J, Rauch SA. et al. In utero and childhood polybrominated diphenyl ether (PBDE) exposures and neurodevelopment in the CHAMACOS study. Environ Health Perspect. 2013;121:257-62
32. Cowell WJ, Margolis A, Rauh VA. et al. Associations between prenatal and childhood PBDE exposure and early adolescent visual, verbal and working memory. Environ Int. 2018;118:9-16
33. Wu HJ, Pu JL, Krafft PR. et al. The molecular mechanisms between autophagy and apoptosis: potential role in central nervous system disorders. Cell Mol Neurobiol. 2015;35:85-99
34. Jiang C, Zhang S, Liu H. et al. The role of the IRE1 pathway in PBDE-47-induced toxicity in human neuroblastoma SH-SY5Y cells in vitro. Toxicol Lett. 2012;211:325-33
35. Pereira LC, Duarte FV, Varela AT. et al. An autophagic process is activated in HepG2 cells to mediate BDE-100-induced toxicity. Toxicology. 2017;376:59-65
36. Pereira LC, Duarte FV, Varela A. et al. Exposure to BDE-153 induces autophagy in HepG2 cells. Toxicol in Vitro. 2017;42:61-68
37. Matus S, Valenzuela V, Hetz C. A new method to measure autophagy flux in the nervous system. Autophagy. 2014;10:710-14
38. Marchi S, Corricelli M, Trapani E. et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med. 2015;7:1403-17
39. Mizushima N, Noda T, Yoshimori T. et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395-98
40. Sun W, Du L, Tang W. et al. PBDE-209 exposure damages learning and memory ability in rats potentially through increased autophagy and apoptosis in the hippocampus neuron. Environ Toxicol Pharmacol. 2017;50:151-58
41. Wang Q, Wei LW, Zhou WT. et al. PCB28 and PCB52 induce hepatotoxicity by impairing the autophagic flux and stimulating cell apoptosis in vitro. Toxicol Lett. 2018;289:28-41
42. You M, Savaraj N, Kuo MT. et al. TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol Cell Biochem. 2013;374:181-90
43. Tsapras P, Nezis IP. Caspase involvement in autophagy. Cell Death Differ. 2017;24:1369-79
44. Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. 2009;122:2554-66
Author contact
![]() Corresponding authors: Dr. Shun Zhang, Phone: 86-27-83691030, Fax: 86-27-83692701, Email: shunzhangedu.cn. Dr. Aiguo Wang, Phone: 86-27-83691030, Fax: 86-27-83692701, Email: wangaiguotjmu.edu.cn
Corresponding authors: Dr. Shun Zhang, Phone: 86-27-83691030, Fax: 86-27-83692701, Email: shunzhangedu.cn. Dr. Aiguo Wang, Phone: 86-27-83691030, Fax: 86-27-83692701, Email: wangaiguotjmu.edu.cn