Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2020; 10(22):9913-9922. doi:10.7150/thno.46417 This issue Cite
Research Paper
Inhibition of Pendrin by a small molecule reduces Lipopolysaccharide-induced acute Lung Injury
Eun Hye Lee1, Mi Hwa Shin2, Mia Gi3,4, Jinhong Park5,6, Doona Song7, Young-Min Hyun8, Ji-Hwan Ryu9,10, Je Kyung Seong11,12, Yoon Jeon13, Gyoonhee Han5,7, Wan Namkung5,6 ![]() , Moo Suk Park2
, Moo Suk Park2 ![]() , Jae Young Choi3,4
, Jae Young Choi3,4 ![]()
1. Division of Pulmonology, Allergy and Critical Care Medicine, Department of Internal Medicine, Yongin Severance Hospital, Yonsei University College of Medicine, Yongin-si, Gyeonggi-do, Republic of Korea.
2. Division of Pulmonology, Department of Internal Medicine, Institute of Chest Diseases, Severance Hospital, Yonsei University College of Medicine, Seoul, South Korea.
3. Department of Otorhinolaryngology, Yonsei University College of Medicine, Seoul, South Korea.
4. The Airway Mucus Institute, Yonsei University College of Medicine, Seoul, South Korea.
5. College of Pharmacy, Yonsei Institute of Pharmaceutical Sciences, Yonsei University, Incheon, South Korea.
6. Department of Integrated OMICS for Biomedical Science, Yonsei University, Seoul, South Korea.
7. Translational Research Center for Protein Function Control, Department of Biotechnology, Yonsei University, Seoul, South Korea.
8. Department of Anatomy, Yonsei University College of Medicine, Seoul, South Korea.
9. Severance Biomedical Science Institute, Yonsei University College of Medicine.
10. Brain Korea 21 PLUS Project for Medical Science, Yonsei University College of Medicine.
11. Laboratory of Developmental Biology and Genomics, BK21 Plus Program for Advanced Veterinary Science and Research Institute for Veterinary Science, College of Veterinary Medicine, and Korea Mouse Phenotyping Center, Seoul National University, Seoul, South Korea.
12. Interdisciplinary Program for Bioinformatics, Seoul National University, Seoul, South Korea.
13. Research Institute, National Cancer Center, South Korea.
*These authors contributed equally to this work.
Received 2020-3-27; Accepted 2020-7-27; Published 2020-8-7
Abstract

Rationale: Pendrin is encoded by SLC26A4 and its mutation leads to congenital hearing loss. Additionally, pendrin is up-regulated in inflammatory airway diseases such as chronic obstructive pulmonary disease, allergic rhinitis, and asthma. In this study, the effects of a novel pendrin inhibitor, YS-01, were investigated in an LPS-induced acute lung injury (ALI) mice model, and the mechanism underlying the effect of YS-01 was examined.
Methods: Lipopolysaccharide (LPS, 10 mg/kg) was intranasally instilled in wild type (WT) and pendrin-null mice. YS-01 (10 mg/kg) was administered intra-peritoneally before or after LPS inhalation. Lung injury parameters were assessed in the lung tissue and bronchoalveolar lavage fluid (BALF). Pendrin levels in the BALF of 41 patients with acute respiratory distress syndrome (ARDS) due to pneumonia and 25 control (solitary pulmonary nodule) patients were also measured.
Results: LPS instillation induced lung injury in WT mice but not in pendrin-null mice. Pendrin expression was increased by LPS stimulation both in vitro and in vivo. YS-01 treatment dramatically attenuated lung injury and reduced BALF cell counts and protein concentration after LPS instillation in WT mice. Proinflammatory cytokines and NF-κB activation were suppressed by YS-01 treatment in LPS-induced ALI mice. In BALF of patients whose ARDS was caused by pneumonia, pendrin expression was up-regulated compared to that in controls (mean, 24.86 vs. 6.83 ng/mL, P < 0.001).
Conclusions: A novel pendrin inhibitor, YS-01, suppressed lung injury in LPS-induced ALI mice and our data provide a new strategy for the treatment of inflammatory airway diseases including sepsis-induced ALI.
Keywords: pendrin, inhibitor, SLC26A4, ALI, ARDS
Introduction
Acute lung injury (ALI), a common and severe pulmonary complication in critical illness, affects approximately 10 to 15% of patients hospitalized in the intensive care unit (ICU) [1]. Acute respiratory distress syndrome (ARDS), the most severe form of ALI, has a mortality rate of approximately 40%, despite modern ICU care [2,3]. Despite decades of research, treatment options for ARDS are limited and supportive care with mechanical ventilation remains the mainstay of ARDS management [4]. ARDS/ALI are characterized by the abrupt onset of hypoxemia with diffuse pulmonary infiltrates, and the accumulation of a protein-rich pulmonary edema that causes a reduction in lung compliance, alveolar collapse, and ventilation-perfusion mismatch [5]. Recent reports suggest that ion pump and channel functions are affected early in sepsis-induced ARDS [6] and that airway epithelial cells may be a valuable therapeutic target for ALI/ARDS treatment.
Pendrin is encoded by SLC26A4 and acts as an anion exchanger that import Cl- and export bases such as HCO3-, I-, OH-, SCN-, and formate [7-10]. Pendrin is expressed in the inner ear and thyroid, and its mutation is associated with prelingual deafness (DFNB4) and Pendred syndrome [11,12]. Normal airway epithelium shows negligible pendrin expression. However, the expression of pendrin is strongly up-regulated in inflammatory airway diseases, such as chronic obstructive pulmonary disease, allergic rhinitis, asthma [13,14]; and enforced pendrin expression induces mucus overproduction with neutrophilic infiltration in mice airway epithelial cells [13]. Madeo et al. reported that SLC26A4 mutation correlated with asthma resistance, although their results did not reach statistical significance due to the small number of patients [15]. Pendrin upregulation is also observed in primary airway epithelial cells when they are cultured with IL-4, IL-13, and IL-17A [8,13,16]. Nakagami et al. reported that pendrin-deficient mice had less allergen-induced airway hyper-reactivity and inflammation compared to control mice, and pendrin mRNA expression in human nasal epithelial cells increased in cases of common cold caused by rhinovirus [17]. Interestingly, pendrin-null mice show reduced lung inflammation in response to Bordetella pertussis [18]. This evidence indicates that pendrin is a critical protein in the pathogenesis of inflammatory airway disease. Recently, Dr. Verkman and his colleagues screened small molecules for pendrin inhibitors and showed that several pendrin inhibitor compounds significantly increased airway surface liquid thickness in cystic fibrosis patient bronchial epithelial cells expressing high levels of pendrin [19]. We also previously performed a cell-based high throughput screening for the identification of small-molecule pendrin inhibitors; the pendrin inhibitor we screened (YS-01) showed a strongly therapeutic effect in an OVA-induced allergy asthma murine model, where it inhibited pendrin/OSCN-/NF-κB-mediated airway inflammation [20]. Recently obtained data show that pendrin is also expressed in alveolar epithelia, and that administration of a non-specific anion exchanger inhibitor (methazolamide) attenuates the LPS-induced ALI phenotype [21].
Thus, we investigated whether the pendrin inhibitor YS-01 showed a therapeutic effect in an LPS-induced ALI mouse model and also examined the mechanism underlying the effects of YS-01. The findings of this study may provide a novel strategy for the treatment of inflammatory airway diseases including sepsis-induced acute lung injury.
Materials and Methods
Experimental animals
Wild-type male C57BL/6J mice, 8-10 weeks of age and weighing 20- 24 g, were purchased from Orient Bio (Sungnam, Republic of Korea). Pendrin knock-out (KO) mice used in the experiment were provided by JY Choi, and transgenic NF-κB reporter and SPC-Cre-ERT2 mice used for in vivo optical imaging (IVIS) were provided by KT Nam and BC Cho of Yonsei university (Table S1 and Figure S1).
LPS-induced ALI in a murine model
LPS (Escherichia coli, O111: B4, Sigma) (10 mg/kg) in 50 μL PBS was administered by intranasal (i.n.) inhalation. The control group was given 50 μL of sterile PBS intranasally. For the pre-treatment model, YS-01 (10 mg/kg) in 50 μL DMSO was administered intra-peritoneally (i.p) 1 h before LPS inhalation. For the post-treatment model, two doses of YS-01 at 6 and 12 h after LPS inhalation were administered. The mice in the pretreatment group were euthanized and their lungs harvested 48 h after LPS inhalation. In the post-treatment group, euthanasia and sample collection occurred 24 h after the LPS administration. For the SCN- experiment, 50 μL of NaOH, NaHCO3, or NaSCN (100 mM) were administered intranasally after YS-01 treatment. PBS was administered in the same manner for the control group. Additional details on the methods for animal experiments are provided in an online data supplement.
Human bronchoalveolar lavage fluid collection
Forty-one patients with ARDS caused by pneumonia who underwent a bronchoalveolar lavage (BAL) were classified as the ARDS group. The 25 patients who were admitted for evaluation of solitary pulmonary nodule (SPN) without evidence of pulmonary infection were classified as the control group at the Severance Hospital between May 2013 and September 2015.
Prior to bronchoscopy, subjects were sedated with midazolam and fentanyl. The bronchoscope was inserted and wedged into the mouth for the BAL. BAL was performed following a standardized protocol (ARDS group: bronchus of pulmonary lesion, Control group: opposite bronchus from lung mass) and 10 cc of BALF was acquired from each patient using approximately 30 mL of 0.9% sodium chloride. Demographic and clinical data, including age, gender, body mass index (BMI), comorbidities, BALF analysis, cause of pneumonia, and final diagnosis were obtained from each participant, as well as medical records.
Statistics
Statistical analysis was performed using Prism 5.0 (GraphPad Software). Group comparisons were performed using a two-tailed Student's t test to compare two groups, and a one-way ANOVA (followed by Bonferroni's multiple comparison post-hoc test) to compare more than two groups. Data are expressed as the mean ± SD. P values of less than 0.05 were considered statistically significant.
Study approval
All animal protocols were approved by the Institutional Animal Care Committee of the Medical College of Yonsei University (2016-0322). Human study protocols were reviewed and approved by the Institutional Review Board of Yonsei University Health Service, Severance Hospital, Seoul, Korea (ARDS group IRB No. 4-2013-0585, control group IRB No. 4-2014-1014). Written informed consent was obtained from patients or their guardians regarding BALF sample use.
Results
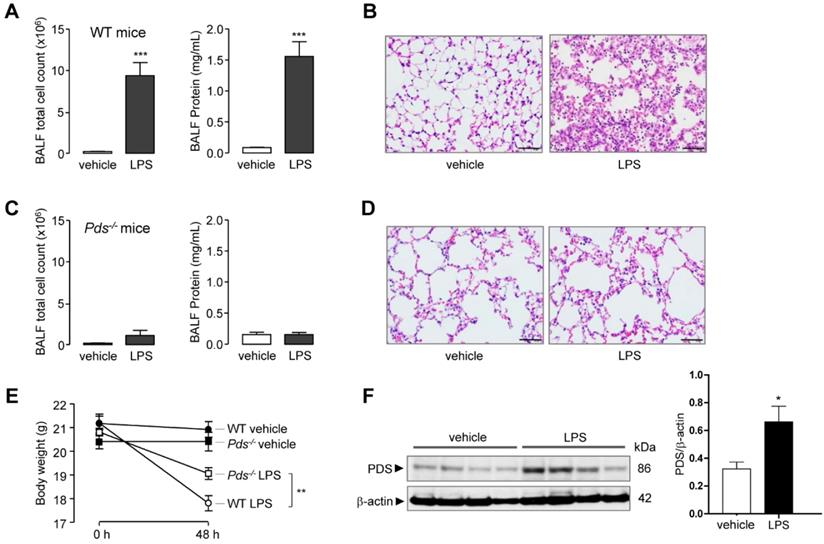
LPS-induced ALI absent in pendrin-null mice
As expected, intranasal LPS instillation induced ALI in WT mice. The total cell count and protein concentration in bronchoalveolar lavage fluid (BALF) was markedly increased after LPS treatment (Figure 1A). Lung histology also showed leukocyte infiltration and lung injury in WT mice (Figure 1B). In contrast, LPS did not increase cell count or protein concentration in pendrin-null mice (Figure 1C). Lung histology also revealed a lack of leukocyte infiltration and lung injury in pendrin-null mice after LPS treatment (Figure 1D). The mean body weight change after 48 hours of LPS instillation was more pronounced in WT mice than in pendrin-null mice (-3.38 g vs. -1.75 g, P < 0.01, Figure 1E). Immunoblot analysis of lung tissue lysates showed that pendrin protein expression was increased after LPS treatment compared to the vehicle control (Figure 1F). These results suggested that pendrin has an essential role in the development of LPS-induced ALI.
Pendrin deficiency attenuated LPS-induced lung injury in mice. Wild type (WT) and pendrin-null (Pds-/-) mice were intranasally administered LPS (10 mg/kg) or vehicle (PBS). (A) Total bronchoalveolar lavage (BAL) cell counts and BAL protein concentration were analyzed 48 h after LPS or PBS administration in WT mice. (B) Representative images of H & E staining of lung tissue 48 h after LPS or PBS administration (×400), scale bars: 50 µm. (C) Total BAL cell counts and BAL protein concentration were analyzed 48 h after LPS or PBS administration in pendrin-null mice. (D) Representative images of H & E staining of lung tissue 48 h after LPS or PBS administration (×400), scale bars: 50 µm. (E) Mice body weight changes. (F) Representative western blot analysis and densitometry of pendrin in lung lysates of LPS untreated and treated WT mice. Data provided are the mean ± SEM (n = 6-8 mice per group), *P < 0.05, **P < 0.01, ***P < 0.001 analyzed by Student's unpaired two-tailed t test.
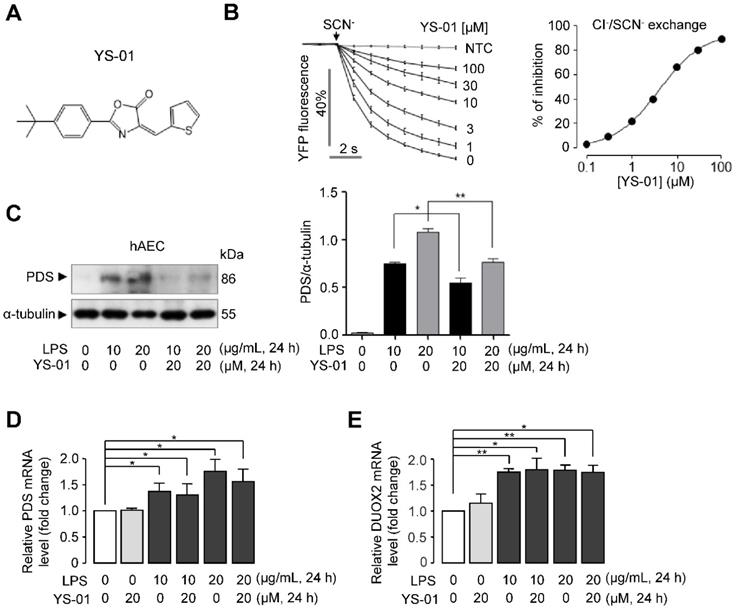
The effect of a novel pendrin inhibitor (YS-01) in human alveolar epithelial cells
In our previous study [20], a novel pendrin inhibitor, YS-01 was identified by the high-throughput screening of 54,400 synthetic compounds. YS-01 potently inhibited Cl-/I-, Cl-/SCN-, Cl-/HCO3-, and Cl-/OH- exchange activity in naso-tracheal epithelia [20]. In this study, we showed that YS-01 potently inhibited the Cl-/SCN- exchange activity of pendrin (IC50 = 4.7 ± 0.82 μM) in pendrin-transfected human alveolar epithelial cells (hAEC) in a dose-dependent manner (Figure 2B). Our previous study showed that long-term treatment of YS-01 significantly reduced the protein expression level of pendrin without changing the mRNA expression level of pendrin [20]. We also investigated the effect of LPS and YS-01 on protein and mRNA levels of pendrin in hAEC. Western blot analysis and densitometry showed that pendrin expression was increased after LPS treatment and suppressed after YS-01 treatment (Figure 2C). Meanwhile, LPS treatment significantly increased mRNA levels of pendrin, but YS-01 did not alter the mRNA levels of pendrin in hAEC (Figure 2D). Previous studies have shown that upregulation of pendrin can activate NF-κB by increasing hypothiocyanite (OSCN-) production via upregulation of dual oxidase (Duox1/Duox2) in airway epithelium of allergic inflammation [22]. Real-time PCR analyses showed that LPS treatment significantly increased the mRNA levels of Duox2, and YS-01 did not alter mRNA levels of Duox2 (Figure 2E).
Novel pendrin inhibitor (YS-01) blocked pendrin activity in human alveolar epithelial cells. (A) Chemical structure of YS-01 pendrin inhibitor. (B) Inhibitory effect of YS-01 on human wild-type pendrin-mediated Cl-/SCN- exchange activity in hAEC expressing human pendrin (mean ± SEM., n = 10). Indicated concentrations of YS-01 were pretreated for 10 min. Dose-response summary (right). (C) Representative western blot analysis of pendrin (PDS) in human alveolar epithelial cells (hAEC) and relative band intensity (mean ± SEM., n = 3). (D) PDS mRNA levels were determined by real-time quantitative PCR in hAEC (mean ± SEM., n = 3). (E) DUOX2 mRNA levels were determined by real-time quantitative PCR in hAEC (mean ± SEM., n = 3~4). *P < 0.05, **P < 0.01 analyzed by Student's unpaired two-tailed t-test.
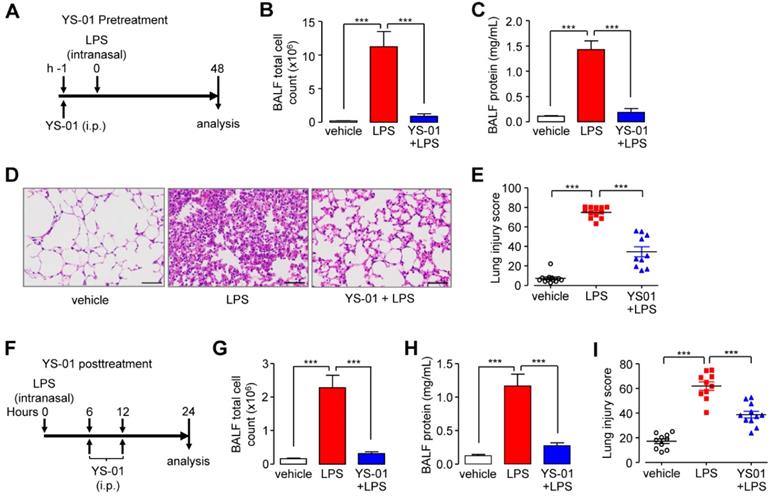
Pendrin inhibition attenuated LPS-induced ALI in mice
To investigate the protective function of YS-01 in an LPS-induced ALI mouse model, mice were treated with YS-01 one hour before LPS intranasal instillation and euthanized 48 hours after LPS administration (Figure 3A). YS-01 (10 mg/kg) pre-treated mice displayed decreased BALF total cell count and protein concentration levels compared to vehicle treated mice (Figures 3B and 3C). YS-01 pre-treatment also significantly reduced the lung injury score compared to vehicle-treated mice, where leukocyte infiltration occurred after LPS exposure (Figures 3D and 3E). To determine whether YS-01 treatment was effective after LPS injury, mice were treated with YS-01 at 6 and 12 h after LPS intranasal instillation, and then euthanized 24 hours after LPS administration (Figure 3F). YS-01 treatment after LPS instillation significantly reduced BALF total cell count and protein concentration, as well as lung injury score, consistent with the results of the YS-01 pre-treatment experiment (Figures 3G-I).
YS-01 suppressed the LPS-induced acute lung injury phenotype in mice. (A) YS-01 (10 mg/kg) was intraperitoneally injected 1 h before LPS treatment. (B) BALF total cell count. (C) BALF protein concentration. (D) Representative images of H&E lung tissue staining (×400), scale bars: 50 µm. (E) Lung injury scores. (F) YS-01 (10 mg/kg) was intraperitoneally injected at 6 and 12 h after LPS inhalation. (G) BALF total cell count. (H) BALF protein concentration. (I) Lung injury scores. Data provided are the mean ± SEM (n = 10-12 mice per group), ***P < 0.001, analyzed by one-way ANOVA with Bonferroni's post hoc test.
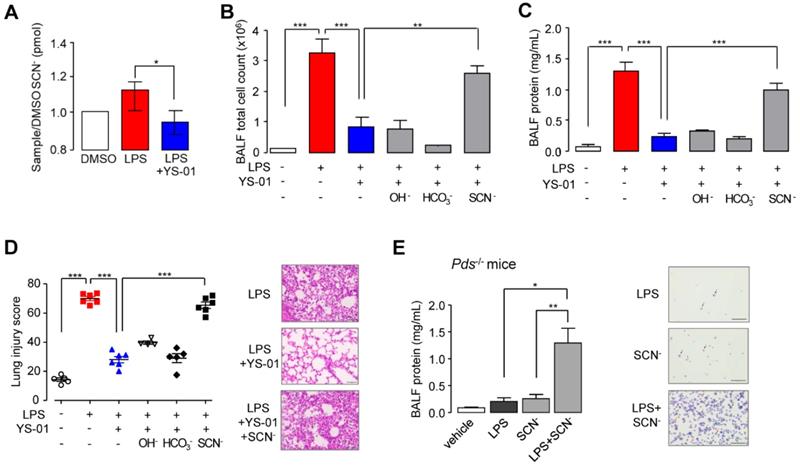
SCN- instillation triggered LPS-induced ALI in the presence of a pendrin inhibitor and in pendrin-null mice
To measure SCN- transport, we used human nasal epithelial (HNE) cells using air-liquid interface culture. The SCN- concentration at the apical surface was increased by LPS exposure and inhibited by YS-01 significantly (Figure 4A). To identify the underlying therapeutic effect mechanism of YS-01 in LPS-induced ALI, we supplied the anions secreted by pendrin (OH-, HCO3-, and SCN-). Intranasal application of NaSCN (50 μL of 100 mM) blocked the protective effects of YS-01 in LPS-induced ALI; BALF total cell count and lung injury score were increased compared to the group treated with LPS alone. However, administration of NaOH and NaHCO3 did not change the effect of YS-01 in LPS-induced ALI mice (Figures 4B and 4C). Histological analysis also revealed that the protective effect of YS-01 on inflammatory cell infiltration and lung injury after LPS administration was abolished by NaSCN administration (Figure 4D). More interestingly, simultaneous application of NaSCN with LPS induced robust lung injury in pendrin-null mice, whereas administration of LPS alone did not induce ALI (Figure 4E). These data strongly indicate that the therapeutic effect of YS-01 results from the SCN- transport function inhibition of pendrin.
SCN- triggered LPS-induced lung injury in the presence of YS-01 or pendrin-null mice. (A) Effect of YS-01 on SCN- transport in HNE cells (mean ± SEM (n = 5 per group). (B) BALF total cell count. LPS (10 mg/kg, i.n.), YS-01 (10 mg/kg, i.p), NaOH (100 mM, i.n.), NaHCO3 (100 mM, i.n.) and NaSCN (100 mM, i.n.) were treated in WT mice. (C) BALF protein concentration. (D) Lung injury score. Representative lung tissues stained with H & E (right) (×400), scale bars: 50 µm. (E) BALF protein concentration in pendrin-null mice. NaSCN (100 mM, i.n.) was applied to the LPS treated pendrin-null mice. Representative mouse BALF cytospin stained with Diff-Quik Stain. Inflammatory cells, especially neutrophils (red arrows) were increased after LPS + NaSCN exposure compared to LPS or NaSCN alone (right). Black arrows represent macrophages (×200), scale bars: 100 µm. Data provided are the mean ± SEM (n = 5-6 per group), *P < 0.05, **P < 0.01, ***P < 0.001, analyzed by one-way ANOVA with Bonferroni's post hoc test.
Pendrin inhibitor blocked the NF-kB pathway and decreased inflammatory cytokines in an LPS-induced ALI mouse model
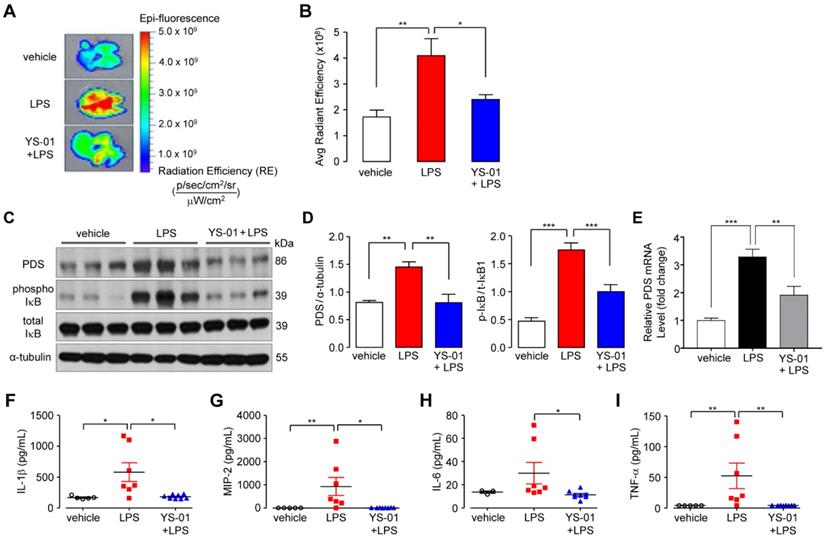
We further dissected the signaling pathway by which YS-01 acts in an LPS-induced ALI model using NF-κB reporter/SPC-Cre-ERT2 mice. Quantitative fluorescence was determined using in vivo optical imaging (IVIS) images of mouse lungs that were aseptically removed immediately prior to imaging. Fluorescence in the excised lungs was increased after LPS treatment, which was suppressed by YS-01 (Figures 5A and 5B). These results suggested that NF-κB activation in LPS instilled mice was suppressed by YS-01. Immunoblot analysis showed that the NF-κB pathway was associated with YS-01 action. Phospho-IκB protein expression, which represented NF-κB activation, was increased after LPS administration, and YS-01 treatment before LPS significantly reduced phospho-IκB expression (Figures 5C and 5D). Pendrin mRNA levels significantly increased after LPS and were decreased by YS-01 (Figure 5E). The levels of cytokines including IL-1ß, tumor necrosis factor-α, and macrophage inflammatory protein (MIP)-2 were significantly increased after LPS administration compared to PBS (Figures 5F-I). IL-6 tended to be increased, relative to PBS, although the difference was statistically insignificant. In contrast, levels of pro-inflammatory cytokines decreased in YS-01 pre-treated mice compared to those that received vehicle (DMSO) treatment after LPS administration (Figures 5F-I).
YS-01 blocked the NF-κB pathway and reduced the levels of proinflammatory cytokines in LPS-induced acute lung injury. (A) Representative images of lungs from NF-κB/SPC-Cre mice exposed to LPS (10 mg/kg) and treated with either YS-01 (10 mg/kg) or a vehicle. IVIS image fluorescence is presented as the radiant efficiency. (B) Average fluorescence was quantified by region of interest analysis using Living Image software. Data provided are the mean ± SEM (n = 9-10 mice per group). (C) Representative western blot analysis in lung lysate. (D) Relative protein levels were measured by densitometry for pendrin and phospho-IκB. (means ± SEM, n = 6 per group). (E) Pendrin mRNA levels were determined by real-time quantitative PCR in lung tissue (means ± SEM, n = 11 per group). (F-I) IL-1β, CXCL2/MIP-2, IL-6 and TNF-α levels were measured by ELISA in lung tissue lysates. Data provided are the mean ± SEM (n = 7-8 mice per group), *P < 0.05, **P < 0.01, **P < 0.001, analyzed by one-way ANOVA with Bonferroni's post hoc test.
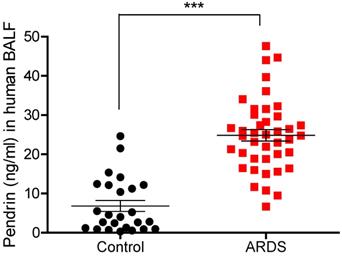
Pendrin levels in human BALF were increased in patients with pulmonary infection
To translate in vitro and in vivo findings to human disease, we measured BALF pendrin protein expression in patients with ARDS caused by pneumonia (ARDS group, n = 41) and patients with a solitary pulmonary nodule (SPN) but no infection (control group, n = 25). Patient clinical characteristics are shown in Table 1. Mean age was not significantly different between the control and ARDS groups (63.8 vs. 65.9, p = 0.517) and males were predominant in both groups (80% vs. 78%, p = 0.851). Among the ARDS patients, the median length of hospital stay was 36 days, and the 28-day mortality rate was 24.4% (Table 1). Detailed clinical information of study patients can be found online in Table S2. Pendrin level was significantly elevated in the BALF of ARDS patients (n = 41) compared to that of the control subjects (n = 25) (mean, 24.86 vs. 6.83 ng/mL, P < 0.001) (Figure 6).
Clinical characteristics of study patients
| Control (n = 25) | ARDS* (n = 41) | P | |
|---|---|---|---|
| Age, years, mean ± SD | 63.8 ± 9.7 | 65.9 ± 13.6 | 0.517 |
| Gender, male, N (%) | 20 (80.0) | 32 (78.0) | 0.851 |
| BMI (kg/m2), mean ± SD | 24.5 ± 4.3 | 22.6 ± 3.0 | 0.040 |
| ICU admission, N (%) | 0 | 41 (100) | - |
| Intubation/ARDS, N (%) | 0 | 41 (100) | - |
| P/F ratio, mean ± SD | - | 157.3 ± 52.8 | - |
| Bacteremia, N (%) | 0 | 9 (22.0) | - |
| Length of stay, d, median (IQR) | 2 (1-2) | 36 (26-57) | - |
| 28-day mortality, N (%) | 0 | 10 (24.4) | - |
| In-hospital mortality, N (%) | 0 | 28 (68.3) | - |
| Pendrin level, ng/mL, mean ± SD | 6.83 ± 6.91 | 24.86 ± 9.28 | <0.001 |
Values are presented as the mean±SD, median (interquartile range, IQR), or number (%);
*ARDS due to pneumonia;
Abbreviations: BMI, body mass index; ICU, intensive care unit; ARDS, acute respiratory distress syndrome; P/F, PaO2/FIO2.
Pendrin levels of human BALF. Patients with ARDS caused by pneumonia displayed increased pendrin levels compared to the control patients (without infection). Pendrin levels were measured from human BALF supernatant by ELISA (Control n = 25, ARDS n = 41). *P < 0.05, **P < 0.01, ***P < 0.001, analyzed by Student's unpaired two-tailed t test.
Discussion
Emerging evidence strongly suggests that pendrin is a key protein in the development of airway inflammatory diseases including asthma, chronic obstructive pulmonary disease, and rhinitis [13,23]. We demonstrated that the pendrin expression level in LPS-treated mouse airways increased. Moreover, we showed that LPS-induced ALI did not develop in pendrin null mice, which strongly indicated the critical role of pendrin in ALI pathogenesis. This is consistent with a recent report that showed pendrin expression was enhanced in LPS-induced ALI, and a non-specific pendrin inhibitor attenuated ALI in mice [21]. This evidence encouraged us to develop a pendrin inhibitor as a novel drug for ALI treatment. We screened more than 54,400 small molecules and found a specific pendrin inhibitor (YS-01) that did not affect other ion transports, such as Cystic fibrosis transmembrane conductance regulator (CFTR) and calcium-activated chloride channel (CaCC) [20]. Pendrin expression was upregulated by LPS treatment in human alveolar epithelial cell culture, which was effectively suppressed by YS-01. Figures 2C-2D show that treatment of YS-01 significantly reduced the protein expression of pendrin without changing mRNA expression in hAEC. These results suggested that YS-01 decreased the protein stability of pendrin and consistent with our previous results in human nasal epithelial cells [20]. Interestingly, YS-01 reduced both protein and mRNA levels of pendrin in the animal model of LPS-induced lung injury (Figures 5D and 5E). The reduction of pendrin mRNA expression levels in YS-01-treated mice may be due to the decreased inflammatory signals by inhibition of pendrin. Decreased pendrin mRNA expression and destabilization of pendrin protein by YS-01 may enhance the reduction of pendrin protein in vivo. Surprisingly, YS-01 almost completely prevented the development of LPS-induced ALI in mice. Furthermore, administration of YS-01 after LPS treatment attenuated lung injury in mice, which indicated that the clinical therapeutic window of pendrin inhibitors is wide enough to include post-ALI periods. We demonstrated that pendrin expression in BALF from pneumonia patients and LPS-treated mouse airways increased, which strongly suggested a high possibility for the clinical application of pendrin inhibitors in inflammatory airway disease.
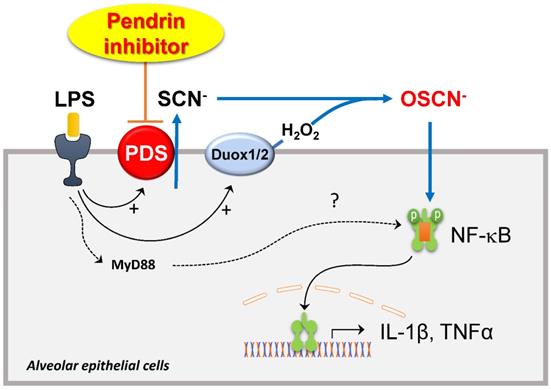
The role of pendrin and the mechanism underlying the therapeutic effects of YS-01 in the ALI model are unclear. We focused on the Cl-/SCN- exchange activity of pendrin and hypothiocyanite (OSCN-), which is synthesized from SCN- transported via several anion transporters (including pendrin) by lactoperoxidases in the airway epithelia. OSCN- is known to be part of an important innate defense system against microbes in the airways [24,25] and also induces airway inflammation in airway epithelia. Recent studies show that IL-4 upregulates the Cl-/SCN- exchange activity of pendrin and increases OSCN- production, which results in NF-κB activation and induces airway inflammation in a murine allergic asthma model [22,26]. We showed that the therapeutic effect of YS-01 on lung injury disappeared when NaSCN was added into the airways of mice. NaSCN application also induced lung injury even in pendrin null mice, whereas LPS-induced ALI had not developed. These data indicate that airway surface SCN- transported by pendrin is an essential component for LPS- induced airway inflammation. NF-κB is a crucial transcription factor for inflammatory responses in airways [27,28] and its inhibition attenuates ALI in vivo [29,30]. We also observed that YS-01 inhibited LPS-induced NF-κB activation and subsequent cytokine production in a murine ALI model and alveolar epithelia. Collectively, our data indicated that the pendrin inhibitor mode of action resulted from YS-01 blocking the transepithelial transport of SCN-, and subsequently inhibiting OSCN- generation and NF-κB activation. This resulted in the suppression of proinflammatory cytokine production (Figure 7). This is a very similar mode of action to that uncovered in our asthmatic mouse model, where pendrin inhibitors attenuated OVA-induced allergic airway inflammation by inhibiting the pendrin/OSCN-/NF-κB cascade [20]. However, if we take into account the previous reports [31,32], which showed that LPS can also activate NF-κB via the TLR4/MyD88 pathway, the reason for YS-01 almost completely suppressing LPS-induced ALI remains unknown. However, the deficiency of ALI phenotypes triggered by NaSCN in pendrin null mice strongly indicates that pendrin-mediated OSCN- dominantly activates the NF-κB cascade in an LPS-induced ALI model.
Schematic diagram of the roles of pendrin and its inhibitor in LPS-induced lung injury. SCN- is actively transported into pulmonary lumens via pendrin at the apical surface of the alveolar epithelia. SCN-, together with H2O2, is catalyzed into OSCN- by peroxidases. The produced OSCNˉ activates NF-κB and causes inflammatory cytokine release, neutrophil infiltration, and subsequent lung injury. Pendrin inhibitor YS-01 blocks the transepithelial transport of SCN- that inhibits the OSCN- induced NF-κB activation and subsequent onset of ALI.
Although critical care for ALI patients has improved, ALI/ARDS mortality remains high and there are limited options for the medical treatment of ALI/ARDS. Because YS-01 showed a strong therapeutic effect on our ALI murine model, pendrin could be a novel target for the medical treatment of ALI/ARDS. It is promising that pendrin expression was upregulated in pneumonia patient BALF, which increases the potential clinical therapeutic benefit of a pendrin inhibitor for ALI/ARDS. However, there are still several issues requiring resolution before the clinical application of pendrin as a drug for ALI/ARDS treatment. First of all, the systemic adverse effect of pendrin inhibitors must be ruled out in areas where pendrin is expressed, including the inner ear, thyroid, and kidney. Although we can rule out hearing loss and hypothyroidism in a mouse model based on our preliminary studies [20], we must examine possible systemic side effects more thoroughly. We can also avoid potential systemic side effects by the local administration of pendrin inhibitors in patients with ALI. Another important issue for the development of pendrin inhibitors as a drug for ALI treatment is the specificity of YS-01 on pendrin. Our previous study [20] showed that YS-01 weakly stimulates SLC26A3 (DRA) and SLC26A6, so it can exert a biological effect in the intestine and kidney. We need to find more specific YS-01 analogues through additional structure-activity relationship analysis. Nevertheless, YS-01 is chemically stable with low cytotoxicity and works at a nanomolar level; it is an excellent compound for the further development of a final candidate for clinical trials.
Although our current study suggests that the inhibition of SCN- transport by pendrin inhibitor is a crucial mechanism, we cannot demonstrate the difference in SCN- concentration in mice airway space. Further study is needed to figure out a proper method to quantify SCN- or OSCN- levels directly. Despite such limitations, we demonstrated that pendrin is essential for LPS-induced ALI, and a small molecule (YS-01) that inhibits pendrin strongly suppressed LPS-induced ALI. Our study indicates that pendrin inhibitors are a promising new drug class for ALI treatment.
Abbreviations
ALI: acute lung injury; ARDS: acute respiratory distress syndrome; BALF: bronchoalveolar lavage fluid; BMI: body mass index; CaCC: calcium-activated chloride channel; CFTR: cystic fibrosis transmembrane conductance regulator; hAEC: human alveolar epithelial cells; ICU: intensive care unit; IVIS: in vivo optical imaging; LPS: Lipopolysaccharide; SPN: solitary pulmonary nodule; WT: wild type.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors thank KT Nam and BC Cho of Yonsei University (South Korea) for their generous donation of Transgenic NF-κB reporter and SPC-Cre-ERT2 mice.
Author contributions
Study Conception and Design, MS park and JY Choi.; Acquisition of Data and experiments, EH Lee, MH Shin, Mia Gi, JH Park, and W Namkung; Analysis and Interpretation of Data, EH Lee, MH Shin, W Namkung, MS Park, and JY Choi; Drafting of Manuscript, EH Lee, W Namkung, MS Park, and JY Choi; Technical Support and consulting, DN Song, YM Hyun, JH Ryu, JK Seoung, Y J, GH Han, W Namkung; All authors read and approved the final manuscript.
Financial/nonfinancial disclosures
This work was supported by grants from the Korea Healthcare Technology R&D Project, Ministry for Health & Welfare Affairs, Republic of Korea (HI08C2149). This work was also supported by the Bio & Medical Technology Development Program of the National Research Foundation of Korea (NRF) funded by the ministry of Science, ICT & Future Planning (NRF-2014M3A9D5A01073865), and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2015R1D1A1A01057695, and NRF-2018R1A6A1A03023718). In addition, this study was supported by a faculty research grant from Yonsei University College of Medicine (6-2018-0164) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP)(2020R1A2C3005787).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goss CH, Brower RG, Hudson LD, Rubenfeld GD. Incidence of acute lung injury in the United States. Crit Care Med. 2003;31:1607-1611
2. Zambon M, Vincent JL. Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest. 2008;133:1120-1127
3. Erickson SE, Martin GS, Davis JL, Matthay MA, Eisner MD. Recent trends in acute lung injury mortality: 1996-2005. Crit Care Med. 2009;37:1574-1579
4. Fan E, Needham DM, Stewart TE. Ventilatory management of acute lung injury and acute respiratory distress syndrome. JAMA. 2005;294:2889-2896
5. Ranieri VM, Rubenfeld GD, Thompson BT. et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526-2533
6. Mutlu GM, Sznajder JI. Mechanisms of pulmonary edema clearance. Am J Physiol Lung Cell Mol Physiol. 2005;289:L685-695
7. Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP. The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999;21:440-443
8. Pedemonte N, Caci E, Sondo E. et al. Thiocyanate transport in resting and IL-4-stimulated human bronchial epithelial cells: role of pendrin and anion channels. J Immunol. 2007;178:5144-5153
9. Sala-Rabanal M, Yurtsever Z, Berry KN, Brett TJ. Novel Roles for Chloride Channels, Exchangers, and Regulators in Chronic Inflammatory Airway Diseases. Mediators Inflamm. 2015;2015:497387
10. Amlal H, Petrovic S, Xu J. et al. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(-)/HCO3(-) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol. 2010;299:C33-41
11. Everett LA, Glaser B, Beck JC. et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet. 1997;17:411-422
12. Everett LA, Belyantseva IA, Noben-Trauth K. et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153-161
13. Nakao I, Kanaji S, Ohta S. et al. Identification of pendrin as a common mediator for mucus production in bronchial asthma and chronic obstructive pulmonary disease. J Immunol. 2008;180:6262-6269
14. Ishida A, Ohta N, Suzuki Y. et al. Expression of pendrin and periostin in allergic rhinitis and chronic rhinosinusitis. Allergol Int. 2012;61:589-595
15. Madeo AC, Manichaikul A, Pryor SP, Griffith AJ. Do mutations of the Pendred syndrome gene, SLC26A4, confer resistance to asthma and hypertension? J Med Genet. 2009;46:405-406
16. Adams KM, Abraham V, Spielman D. et al. IL-17A induces Pendrin expression and chloride-bicarbonate exchange in human bronchial epithelial cells. PLoS One. 2014;9:e103263
17. Nakagami Y, Favoreto S Jr, Zhen G. et al. The epithelial anion transporter pendrin is induced by allergy and rhinovirus infection, regulates airway surface liquid, and increases airway reactivity and inflammation in an asthma model. J Immunol. 2008;181:2203-2210
18. Scanlon KM, Gau Y, Zhu J. et al. Epithelial anion transporter pendrin contributes to inflammatory lung pathology in mouse models of Bordetella pertussis infection. Infect Immun. 2014;82:4212-4221
19. Haggie PM, Phuan PW, Tan JA, Zlock L, Finkbeiner WE, Verkman AS. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J. 2016;30:2187-2197
20. Park J, Lee HJ, Song D. et al. Novel pendrin inhibitor attenuates airway hyperresponsiveness and mucin expression in experimental murine asthma. J Allergy Clin Immunol. 2019;144:1425-1428.e12
21. Jia CE, Jiang D, Dai H, Xiao F, Wang C. Pendrin, an anion exchanger on lung epithelial cells, could be a novel target for lipopolysaccharide-induced acute lung injury mice. Am J Transl Res. 2016;8:981-992
22. Suzuki S, Ogawa M, Ohta S. et al. Induction of Airway Allergic Inflammation by Hypothiocyanite via Epithelial Cells. J Biol Chem. 2016;291:27219-27227
23. Yick CY, Zwinderman AH, Kunst PW. et al. Transcriptome sequencing (RNA-Seq) of human endobronchial biopsies: asthma versus controls. Eur Respir J. 2013;42:662-670
24. Chandler JD, Min E, Huang J, Nichols DP, Day BJ. Nebulized thiocyanate improves lung infection outcomes in mice. Br J Pharmacol. 2013;169:1166-1177
25. Chandler JD, Min E, Huang J. et al. Antiinflammatory and Antimicrobial Effects of Thiocyanate in a Cystic Fibrosis Mouse Model. Am J Respir Cell Mol Biol. 2015;53:193-205
26. Izuhara K, Suzuki S, Ogawa M. et al. The Significance of Hypothiocyanite Production via the Pendrin/DUOX/Peroxidase Pathway in the Pathogenesis of Asthma. Oxid Med Cell Longev. 2017;2017:1054801
27. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7-11
28. Kim SR, Kim HJ, Kim DI. et al. Blockade of Interplay between IL-17A and Endoplasmic Reticulum Stress Attenuates LPS-Induced Lung Injury. Theranostics. 2015;5:1343-1362
29. Brasier AR. Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease. Expert Rev Respir Med. 2018;12:931-939
30. Liu L, Zhou X, Shetty S, Hou G, Wang Q, Fu J. HDAC6 inhibition blocks inflammatory signaling and caspase-1 activation in LPS-induced acute lung injury. Toxicol Appl Pharmacol. 2019;370:178-183
31. Chuang CY, Chen TG, Tai YT. et al. Toll-like receptor 2-mediated sequential activation of MyD88 and MAPKs contributes to lipopolysaccharide-induced sp-a gene expression in human alveolar epithelial cells. Immunobiology. 2011;216:707-714
32. Feng G, Sun B, Liu HX, Liu QH, Zhao L, Wang TL. EphA2 antagonism alleviates LPS-induced acute lung injury via Nrf2/HO-1, TLR4/MyD88 and RhoA/ROCK pathways. Int Immunopharmacol. 2019;72:176-185
Author contact
![]() Corresponding authors: Wan Namkung, College of Pharmacy, Yonsei University, Veritas hall D, 85 Songdogwahak-ro, Yeonsu-gu, Incheon 21983, South Korea. Tel.: +82-32-749-4519; Fax: +82-32-749-4105; E-mail: wnamkungac.kr; Moo Suk Park, Division of Pulmonology, Department of Internal Medicine, Institute of Chest Diseases, Severance Hospital, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. Tel.: +82-2-2228-1955; Fax: +82-2-393-6884; E-mail: pms70ac; Jae Young Choi, Department of Otorhinolaryngology, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. Tel.: +82-2-2228-3603; Fax: +82-2-393-0580; E-mail: jychoiac.
Corresponding authors: Wan Namkung, College of Pharmacy, Yonsei University, Veritas hall D, 85 Songdogwahak-ro, Yeonsu-gu, Incheon 21983, South Korea. Tel.: +82-32-749-4519; Fax: +82-32-749-4105; E-mail: wnamkungac.kr; Moo Suk Park, Division of Pulmonology, Department of Internal Medicine, Institute of Chest Diseases, Severance Hospital, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. Tel.: +82-2-2228-1955; Fax: +82-2-393-6884; E-mail: pms70ac; Jae Young Choi, Department of Otorhinolaryngology, Yonsei University College of Medicine, 50-1 Yonsei-ro, Seodaemun-gu, Seoul 03722, Republic of Korea. Tel.: +82-2-2228-3603; Fax: +82-2-393-0580; E-mail: jychoiac.